 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Víctor Sánchez-Margalet | + 2548 word(s) | 2548 | 2020-07-07 04:43:10 | | | |
| 2 | Nicole Yin | -1176 word(s) | 1372 | 2020-11-05 09:32:49 | | |
Video Upload Options
Leptin is highly expressed in placenta, mainly by trophoblastic cells, where it has an important autocrine trophic effect. Moreover, increased leptin levels are found in the most frequent pathology of pregnancy: gestational diabetes, where leptin may mediate the increased size of placenta and the fetus, which becomes macrosomic. In fact, leptin mediates the increased protein synthesis observed in trophoblasts from gestational diabetic subjects. In addition, leptin seems to facilitate nutrients transport to the fetus in gestational diabetes by increasing the expression of the glycerol transporter aquaporin-9. The high plasma leptin levels found in gestational diabetes may be potentiated by leptin resistance at central level, and obesity-associated inflammation is playing a role in this leptin resistance. Therefore, the importance of anti-inflammatory nutrients to modify the pathology of pregnancy is clear. In fact, nutritional intervention is the first line approach to the treatment of gestational diabetes mellitus. However, more nutritional intervention studies with some nutraceuticals, such as polyphenols or polyunsaturated fatty acids, or nutritional supplementation with micronutrients or probiotics in pregnant women are needed, in order to achieve a high level of evidence. In this context, Mediterranean diet has been recently found to reduce the risk of gestational diabetes in a multicenter randomized trial. This review will focus on the impact of maternal obesity on placental inflammation and nutrients transport considering the mechanisms by which leptin may influence maternal and fetal health in this setting, as well as its role in pregnancy pathologies
1. Definition
The hormone leptin, discovered in 1994[1], critically regulates body weight and metabolism at central level in the brain[2], and disruption of leptin/leptin receptor (LEPR) signaling results in morbid obesity and severe metabolic disease[3][4].
2. Introduction
In individuals of normal weight, the brain responds to increased plasma leptin levels by reducing food intake and increasing energy expenditure[5][6]. Leptin and leptin receptors are highly expressed in the preoptic area (POA), in the arcuate nucleus (ARC) of the hypothalamus as well as in other regions, such as the lateral hypothalamus, ventromedial hypothalamus and dorsomedial hypothalamus (DMH) [7]. There, it regulates energy homoeostasis and the neuroendocrine function, among other functions[8]. In these regions, leptin signaling is mediated by the JAK2/Stat3 pathway, in which several negative regulators of JAK2, including SOCS3 and PTP1B, have been reported to promote obesity[9][10], supporting the notion that JAK2 inhibitory molecules increase risk for leptin resistance and obesity. Therefore, hyperleptinemia and hypothalamic inflammation in diet-induced obesity may activate a common negative regulator of leptin signaling, SOCS3 or PTP1B, and contribute to central leptin resistance. In fact, up-regulation of SOCS3 in proopiomelanocortin (POMC) neurons leads to impairment of STAT3 signaling, with consequential leptin resistance and obesity, as well as glucose intolerance[11]. It has also been reported that mice with whole body or neuron-specific deletion of PTP1B are hypersensitive to leptin, and are resistant to diet-induced obesity[12]. Importantly, obesity is associated with impaired adipose sympathetic nerve transmissions[13][14], but the underlying mechanism is poorly understood. In this context, the leptin resistance at a central level may prevent negative feedback on the anti-inflammatory action of the sympathetic nervous system (SNS)[4][15]. That is why leptin is now considered one of the adipokines responsible for the inflammatory state found in obesity that could predispose to GDM. Surprisingly, Sh2b1 (an SH2 and PH domain-containing adaptor protein)[16][17] has emerged as an endogenous sensitizer for leptin action on the sympathetic nervous system (SNS) and energy expenditure, perhaps by enhancing JAK2 activation[18]. In this way, the LepR Sh2b1 neuron mediates leptin stimulation of the SNS and supports the preservation of adipose SNS against degeneration[19].
Apart from the JAK-2/Stat-3 pathway, activation of the MC4R signaling pathway by proopiomelanocortin (POMC)-derived melanocyte stimulating hormone (MSH) peptides also represents a critical convergence point in the control of body weight. The leptin–melanocortin pathway (MC4R pathway) integrates parallel inputs from the orexigenic peptides ghrelin, neuropeptide Y (NPY) and agouti-related peptide (AgRP), and activation of the MC4R pathway dominantly counteracts these orexigens. Limited efficacy of lifestyle intervention in individuals with mutations in gene-encoding components of this pathway demonstrates its importance in the control of body weight homeostasis[20][21].
Therefore, leptin can act as metabolic switch connecting the nutritional status of the body to high energy-consuming processes. This is especially important in pregnancy, where leptin not only modulates satiety and energy homoeostasis in the mother[2][22], but it is also produced by the placenta, which responds to the environment attempting to maintain fetal viability. This placental production of leptin is one of the major sources of higher levels of maternal circulating leptin other than maternal gain of fat mass[23]. Thus, the effects of placental leptin on the mother may contribute to endocrine-mediated alterations in energy balance, such as the mobilization of maternal fat, which could further aggravate the insulin resistance associated with pregnancy and the onset of GDM[24][25]. In fact, obese pregnant women have significantly elevated plasma leptin concentrations compared with nonobese pregnant women throughout pregnancy[26]. Moreover, maternal obesity is also associated with changes in the placental function and structure, which likely impact fetal growth and development. For example, obesity has been associated with several changes related mainly with placental size, hypervascularization, higher branching capillaries of the villi (chorangiosis)[27][28] and increased glycogen deposits, among others. Increased macrophage infiltration is also evident in the placenta of obese women, suggesting an exaggeration of the inflammatory state which occurs in normal pregnancy[29]. However, it is unclear which histological changes are due to the pathophysiology and which are compensatory adaptations to this disease. Regardless, alterations in placental nutrient and hormone transporter capacity have been demonstrated in human and animal models of obesity, and are hypothesized as a mechanism leading to an accelerated fetal growth trajectory and macrosomia[26]. In this sense, we have demonstrated that the increased expression of aquaporin-9 (AQP9) (or others aquaglyceroporins) observed in placentas from obese women with GDM could be mediated by hyperleptinemia, suggesting an increase in the transport of glycerol to the fetus and thus contributing to the increased energy intake requirements in the macrosomic fetus in GDM[30]. Leptin has also been identified as a critical trophic factor that influences the development of the hypothalamic projections[30]. Alterations in the pattern of leptin secretion (premature peak, excess, or deficiency) during neonatal life could have significant adverse effects on hypothalamic development and metabolic phenotype[31] (Figure 1).
Finally, one of the peripheral functions of leptin is a regulatory role in the interplay between energy metabolism and the immune system, which is, in part, responsible for the inflammatory state associated to obesity[32]. Several inflammatory mediators produced by inflammatory cells also regulate leptin expression and promote the development of chronic inflammation[33]. In this regard, leptin effects include the inflammation and the modulation of innate and adaptive immunity[34][35]. Therefore, proinflammatory leptin actions might also have significant implications in the pathogenesis of GDM[18][36].
Taken all together, since hypothalamic inflammation results in central leptin resistance and hepatic insulin resistance, blocking the peripheral and central inflammation induced by a high fat diet could have the potential to treat obesity and GDM. Therefore, novel therapies incorporating effective natural agents (macro and micronutrients), particularly agents with the dual properties of preventing inflammation and controlling body weight by improving leptin sensitivity, might be an alternative intervention targeting obesity and GDM.
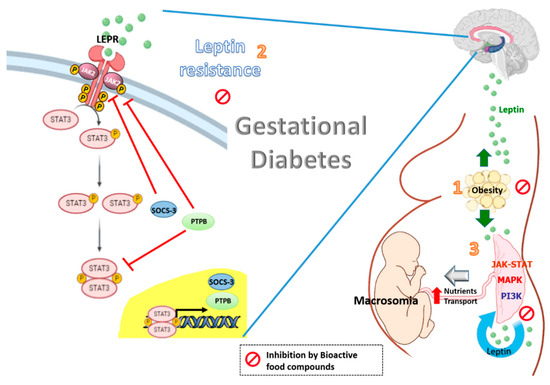
Figure 1. Effects of bioactive food compounds on the leptin resistance associated with obesity and gestational diabetes.
Leptin levels are increased in gestational diabetes with obesity (1). The high plasma leptin levels may be potentiated by leptin resistance at central level, in which SOCS3 and PTPB are induced by leptin and involving in a negative feed-back loop. The resulting effect is a decrease in the leptin-induced activation of the JAK2/STAT-3 signaling, leading to a reduction in the central effects of leptin (2). Leptin also impacts the placenta itself in an autocrine/paracrine fashion. The integration of numerous signaling by intracellular regulatory pathways such as MAPK, PI3K and JAK-STAT has been demonstrated to increase the size of the placenta and to affect placental nutrient transport and fetal growth (macrosomia) (3). Bioactive food compounds such as polyphenols might reduce circulating leptin levels, partly decreasing leptin expression in the placenta from women with GDM. The resulting effect is a decrease in the leptin resistance at a central level and optimal placental nutrients transport.
References
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432, doi:10.1038/372425a0.
- Friedman, J.M.; Halaas, J.L. 1998 Friedman.pdf. Nature 1998, 395, 763–770, doi:10.1038/27376.
- Zhang, Y.; Chua, S. Leptin function and regulation. Compr. Physiol. 2018, 8, 351–369, doi:10.1002/cphy.c160041.
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222.
- Harris, R.B.S.; Apolzan, J.W. Changes in glucose tolerance and leptin responsiveness of rats offered a choice of lard, sucrose, and chow. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1327-39, doi:10.1152/ajpregu.00477.2011.
- van den Heuvel, J.K.; Eggels, L.; van Rozen, A.J.; Luijendijk, M.C.M.; Fliers, E.; Kalsbeek, A.; Adan, R.A.H.; la Fleur, S.E. Neuropeptide Y and leptin sensitivity is dependent on diet composition. J. Neuroendocrinol. 2014, 26, 377–85, doi:10.1111/jne.12155.
- Håkansson, M.L.; Brown, H.; Ghilardi, N.; Skoda, R.C.; Meister, B. Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J. Neurosci. 1998, 18, 559–572, doi:10.1523/jneurosci.18-01-00559.1998.
- Park, H.K.; Ahima, R.S. Leptin signaling. F1000Prime Rep. 2014, 6.
- Matarazzo, V.; Schaller, F.; Nédélec, E.; Benani, A.; Pénicaud, L.; Muscatelli, F.; Moyse, E.; Bauer, S. Inactivation of Socs3 in the hypothalamus enhances the hindbrain response to endogenous satiety signals via oxytocin signaling. J. Neurosci. 2012, 32, 17097–17107, doi:10.1523/JNEUROSCI.1669-12.2012.
- Cheng, A.; Uetani, N.; Simoncic, P.D.; Chaubey, V.P.; Lee-Loy, A.; McGlade, C.J.; Kennedy, B.P.; Tremblay, M.L. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev. Cell 2002, 2, 497–503, doi:10.1016/S1534-5807(02)00149-1.
- Reed, A.S.; Unger, E.K.; Olofsson, L.E.; Piper, M.L.; Myers, M.G.; Xu, A.W. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 2010, 59, 894–906, doi:10.2337/db09-1024.
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917–924, doi:10.1038/nm1435.
- Dodt, C.; Lönnroth, P.; Fehm, H.L.; Elam, M. The subcutaneous lipolytic response to regional neural stimulation is reduced in obese women. Diabetes 2000, 49, 1875–1879, doi:10.2337/diabetes.49.11.1875.
- Coppack, S.W.; Horowitz, J.F.; Paramore, D.S.; Cryer, P.E.; Royal, H.D.; Klein, S. Whole body, adipose tissue, and forearm norepinephrine kinetics in lean and obese women. Am. J. Physiol. - Endocrinol. Metab. 1998, 275, doi:10.1152/ajpendo.1998.275.5.e830.
- Morris, D.L.; Rui, L. Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. - Endocrinol. Metab. 2009, 297, E1247-59.
- Li, Z.; Zhou, Y.; Carter-Su, C.; Myers, M.G.; Rui, L. SH2B1 enhances leptin signaling by both janus kinase 2 Tyr813 phosphorylation-dependent and -independent mechanisms. Mol. Endocrinol. 2007, 21, 2270–2281, doi:10.1210/me.2007-0111.
- Duan, C.; Li, M.; Rui, L. SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. J. Biol. Chem. 2004, 279, 43684–91, doi:10.1074/jbc.M408495200.
- Jiang, L.; Su, H.; Wu, X.; Shen, H.; Kim, M.H.; Li, Y.; Myers, M.G.; Owyang, C.; Rui, L. Leptin receptor-expressing neuron Sh2b1 supports sympathetic nervous system and protects against obesity and metabolic disease. Nat. Commun. 2020, 11, doi:10.1038/s41467-020-15328-3.
- Farooqi, I.S.; O’Rahilly, S. 20 years of leptin: human disorders of leptin action. J. Endocrinol. 2014, 223, T63-70, doi:10.1530/JOE-14-0480.
- Krishna, R.; Gumbiner, B.; Stevens, C.; Musser, B.; Mallick, M.; Suryawanshi, S.; Maganti, L.; Zhu, H.; Han, T.H.; Scherer, L.; et al. Potent and selective agonism of the melanocortin receptor 4 with MK-0493 does not induce weight loss in obese human subjects: energy intake predicts lack of weight loss efficacy. Clin. Pharmacol. Ther. 2009, 86, 659–66, doi:10.1038/clpt.2009.167.
- Houseknecht, K.L.; Portocarrero, C.P. Leptin and its receptors: regulators of whole-body energy homeostasis. Domest. Anim. Endocrinol. 1998, 15, 457–75, doi:10.1016/s0739-7240(98)00035-6.
- Lin, K.C. Increase of maternal plasma leptin concentrations during pregnancy: comparison with nonpregnant women. Kaohsiung J. Med. Sci. 1999, 15, 640–5.
- Higgins, M.; Felle, P.; Mooney, E.E.; Bannigan, J.; McAuliffe, F.M. Stereology of the placenta in type 1 and type 2 diabetes. Placenta 2011, 32, 564–9, doi:10.1016/j.placenta.2011.04.015.
- Jirkovská, M.; Kučera, T.; Kaláb, J.; Jadrníček, M.; Niedobová, V.; Janáček, J.; Kubínová, L.; Moravcová, M.; Zižka, Z.; Krejčí, V. The branching pattern of villous capillaries and structural changes of placental terminal villi in type 1 diabetes mellitus. Placenta 2012, 33, 343–51, doi:10.1016/j.placenta.2012.01.014.
- Roberts, K.A.; Riley, S.C.; Reynolds, R.M.; Barr, S.; Evans, M.; Statham, A.; Hor, K.; Jabbour, H.N.; Norman, J.E.; Denison, F.C. Placental structure and inflammation in pregnancies associated with obesity. Placenta 2011, 32, 247–54, doi:10.1016/j.placenta.2010.12.023.
- Yan, B.; Yu, Y.; Lin, M.; Li, Z.; Wang, L.; Huang, P.; Song, H.; Shi, X.; Yang, S.; Li, X.; et al. High, but stable, trend in the prevalence of gestational diabetes mellitus: A population-based study in Xiamen, China. J. Diabetes Investig. 2019, 10, 1358–1364, doi:10.1111/jdi.13039.
- Vilariño-García, T.; Pérez-Pérez, A.; Dietrich, V.; Fernández-Sánchez, M.; Guadix, P.; Dueñas, J.L.; Varone, C.L.; Damiano, A.E.; Sánchez-Margalet, V. Increased Expression of Aquaporin 9 in Trophoblast from Gestational Diabetic Patients. Horm. Metab. Res. 2016, 48, 535–539, doi:10.1055/s-0042-105152.
- Yura, S.; Itoh, H.; Sagawa, N.; Yamamoto, H.; Masuzaki, H.; Nakao, K.; Kawamura, M.; Takemura, M.; Kakui, K.; Ogawa, Y.; et al. Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab. 2005, 1, 371–8, doi:10.1016/j.cmet.2005.05.005.
- Pérez-Pérez, A.; Vilariño-García, T.; Fernández-Riejos, P.; Martín-González, J.; Segura-Egea, J.J.; Sánchez-Margalet, V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 2017, 35, 71–84.
- Lago, F.; Dieguez, C.; Gómez-Reino, J.; Gualillo, O. The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev. 2007, 18, 313–325.
- Sánchez-Margalet, V.; Martín-Romero, C.; Santos-Alvarez, J.; Goberna, R.; Najib, S.; Gonzalez-Yanes, C. Role of leptin as an immunomodulator of blood mononuclear cells: Mechanisms of action. Clin. Exp. Immunol. 2003, 133, 11–19.
- Martín-Romero, C.; Santos-Alvarez, J.; Goberna, R.; Sánchez-Margalet, V. Human leptin enhances activation and proliferation of human circulating T lymphocytes. Cell. Immunol. 2000, 199, 15–24, doi:10.1006/cimm.1999.1594.
- Pérez-Pérez, A.; Toro, A.; Vilariño-García, T.; Maymó, J.; Guadix, P.; Dueñas, J.L.; Fernández-Sánchez, M.; Varone, C.; Sánchez-Margalet, V. Leptin action in normal and pathological pregnancies. J. Cell. Mol. Med. 2018, 22, 716–727.
- Qiu, C.; Williams, M.A.; Vadachkoria, S.; Frederick, I.O.; Luthy, D.A. Increased maternal plasma leptin in early pregnancy and risk of gestational diabetes mellitus. Obstet. Gynecol. 2004, 103, 519–525, doi:10.1097/01.AOG.0000113621.53602.7a.
- Cai, D.; Liu, T. Hypothalamic inflammation: a double-edged sword to nutritional diseases. Ann. N. Y. Acad. Sci. 2011, 1243.
- SMFM Statement: Pharmacological treatment of gestational diabetes SMFM Publications Committee. Am. J. Obstet. Gynecol. 2018, 218, B2–B4, doi:10.1016/j.ajog.2018.01.041.

