 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simon W Jones | + 2830 word(s) | 2830 | 2021-08-05 12:08:34 | | | |
| 2 | Beatrix Zheng | + 186 word(s) | 3016 | 2021-08-08 04:49:47 | | | | |
| 3 | Beatrix Zheng | + 186 word(s) | 3016 | 2021-08-08 04:50:28 | | | | |
| 4 | Rita Xu | + 200 word(s) | 3216 | 2021-08-23 04:59:00 | | |
Video Upload Options
Oligonucleotide therapeutics represent an emerging but highly promising class of therapeutics to treat inflammatory joint disease. Although yet to be successfully tested in clinical trials for arthritis treatment, data from preclinical experimental models of arthritis provide evidence that the intra-articular delivery of oligonucleotides can modify OA disease pathology, by reducing synovitis, preventing sclerotic bone formation and protecting from cartilage damage. Importantly, since oligonucleotide therapeutics are based on gene sequences, they are expected to act specifically on the target gene, and thus may be considered less likely to have off-target effects and to elicit adverse side effects.
1. Introduction
RA is one of the most common chronic inflammatory conditions, with an estimated prevalence of up to 2% in some ethnic groups [1]. Largely a result of inflammation to the synovial lining (synovitis) of the joint, RA causes joint pain, stiffness, swelling and increased fatigue and results in increased disability and reduced quality of life. Furthermore, around a third of patients are unable to work within 2 years of diagnosis, thus causing a substantial socioeconomic burden [2][3][4]. There are currently several approved pharmacological drugs for the therapeutic management of RA patients [5]. Corticosteroids and non-steroidal anti-inflammatory drugs such as diclofenac can be administered locally to the painful joint by topical application to reduce joint pain and swelling [6]. However, these drugs do not modify the disease process and are also associated with adverse side effects over the long term when taken chronically. For example, glucocorticoids, when taken chronically, can lead to increased bone resorption [7] and skeletal muscle atrophy [8]. Importantly, the aim of current RA therapies is therefore to target the underlying disease pathology, a “treat-to-target” strategy to influence the disease course, not simply to reduce symptoms [9]. To this end, a number of disease-modifying anti-rheumatic drugs (DMARDs) have been approved for clinical use, such as methotrexate, hydroxychloroquine, sulfasalazine and leflunomide, which are immunomodulatory drugs [10]. For example, methotrexate inhibits leukotriene B4 synthesis by neutrophils, and suppresses the production of the pro-inflammatory cytokines IL-1, IL-6 and IL-8 [11][12]. Leflunomide inhibits pyrimidine synthesis, thus reducing lymphocyte proliferation [13], whilst hydroxychloroquine reduces immune responses [14] by inhibiting the toll-like receptor signalling [15]. The precise mechanism of action of sulfasalazine is not fully understood [16], but it is known to inhibit NF-kB activity and thus the production of inflammatory cytokines including tumour necrosis factor alpha (TNF-α) [17]. In recent years, improved understanding of the molecular pathology underlying RA disease has led to a revolution in RA treatment [18], with the emergence of a number of biological drugs designed to target specific disease-associated inflammatory cytokines (most notably TNF-α), and drugs targeting the activity of specific immune cell populations. Monoclonal antibodies targeting TNF-α, including adalimumab (Humira), etanercept and infliximab, have all demonstrated efficacy in reducing disease activity scores (DAS) in patients and in maintaining the disease in remission [19][20][21], as determined by the number of swollen and painful joints and biomarkers of inflammation; thus, reducing the risk of irreversible damage to joints. Similar disease modification has been achieved by biologics targeting other inflammatory immune processes. For example, tocilizumab, a monoclonal antibody targeting the pro-inflammatory IL6 [22], rituximab, a monoclonal antibody targeting CD20 on B cells [23] and abatacept, a fusion protein composed of the Fc region of the immunoglobulin IgG1 fused to the extracellular domain of CTLA-4, which inhibits antigen-presenting cells from providing the co-stimulatory signal required for T-cell activation [24]. However, despite these successes, all these immunomodulatory medications are associated with adverse side effects and an increase in the risk of developing serious infections, such as pneumonia, due to their action in modulating the immune system [25][26]. Furthermore, with chronic treatment, efficacy can be greatly diminished due to the patient developing anti-drug neutralising antibodies [27]. Therefore, there is still a great unmet medical need to develop more effective and safer RA therapeutics.
Similarly to RA, OA results in joint pain and increased stiffness, which leads to progressive disability and a reduced quality of life, amounting to a huge socioeconomic burden. Unfortunately, since age is a significant risk factor for OA, the prevalence of OA is set to increase in developed countries with our increasingly ageing society [28]
Unfortunately, current treatment options for patients are limited. Despite a number of clinical trials, including with biologics targeting pro-inflammatory cytokines such as adalimumab and tocilizumab [29], there are no pharmacological drugs that have been proven to modify the disease course of OA (DMOADs) [30]. Additionally, although platelet-rich plasma and glucosamine-based supplementation show some promise for alleviating pain and delaying OA progression, these effects have been highly variable and debated in trials [31][32]. Therefore, patients are advised to take non-steroidal anti-inflammatory drugs such as ibuprofen, which have limited efficacy and are associated with toxicity over the long term. Patients with knee OA can receive intra-articular corticosteroid injections into the diseased joint to alleviate pain and inflammation [27]. However, the benefit for patients is highly unpredictable, with ~30% of knee OA patients reporting little to no improvement in symptomatic pain [33]. Furthermore, repeated steroid injections into the joint can exacerbate cartilage damage. As a result, OA patients live with painful symptoms for several years before undergoing a joint replacement surgery, a procedure with a high percentage of patient dissatisfaction. A particular challenge in developing a DMOAD has been the effective delivery of drugs into the cartilage, which as an avascular tissue, is not highly amenable for conventional drug delivery. In order to target the chondrocyte cells that mediate cartilage OA pathology, such drugs need to be able to penetrate the full depth of the cartilage tissue [34]. Furthermore, the focus for many years on identifying candidate targets that directly mediate cartilage degradation, such as the matrix metalloproteases (MMPs), neglected the important role of the other tissues in the joint in driving OA disease progression, such as the synovium and subchondral bone. To this end, the development of oligonucleotides, that modulate expression of targets at an upstream level, where functional protein products are not made, may offer the potential to target pathological processes across multiple cell types, including chondrocytes in the cartilage, osteoblasts in the bone and fibroblasts in the synovium.
In this review, we discuss the potential for the development of oligonucleotide therapies in both RA and OA joint disease by examining the evidence for oligonucleotide therapies to modulate disease pathology and disease-associated cellular pathways within the multiple tissues of the joint ( Figure 1 ).
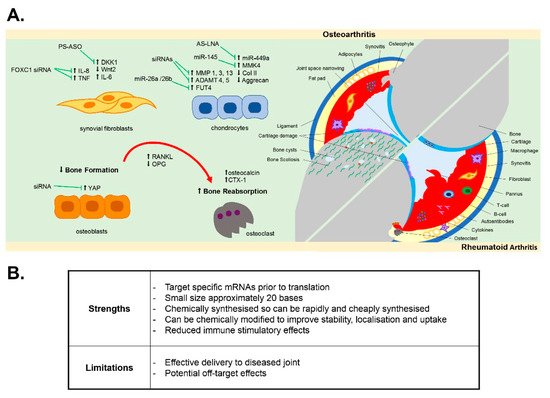
2. Oligonucleotides Targeting Synovial Inflammation
Inflammation of the synovial membrane (synovitis) is a key hallmark of both RA and OA joint disease. Fibroblast-like synoviocytes (FLS), also termed synovial fibroblasts, become activated and hyperplastic in RA [35][36]. Through the infiltration and subsequent interaction of immune cells in the synovium (including leukocytes and resident macrophages), synovial fibroblasts contribute to a sustained chronic inflammatory state within the joint by releasing pro-inflammatory cytokines into the synovial joint fluid, such as TNFα and IL-6 [36][37], which also promotes bone resorption. Synovitis and the associated bone erosion are detected by magnetic resonance imagining (MRI) of the RA joint [38]. Similar to RA, synovitis is now widely recognised to play a significant role in OA joint pathology, with synovitis evidence by ultrasound, MRI and histopathology prior to radiographic signs of cartilage damage, with increased infiltration of activated B and T cells and synovial proliferation and hypertrophy [29][39][40][41][42]. Synovial fibroblasts from OA patients are more inflammatory compared to non-diseased patients and secrete greater levels of pro-inflammatory cytokines IL-6 and IL-8 [43].
In attempting to develop oligonucleotide therapies to target synovitis, modulation of the activated proliferative inflammatory synovial fibroblast phenotype using antisense oligonucleotides has been documented. For example, Nakazawa et al. reported that antisense oligonucleotides targeting Notch-1 protein inhibited both basal and TNFα-induced proliferation of human synovial fibroblasts isolated from either RA or OA patient synovium [44], whilst antisense knockdown of the gene PTPN11, which encodes SHP-2 (a known proto-oncogene), was reported to inhibit migration and survival of RA synovial fibroblasts [45]. More recently, susceptibility of human OA and RA synovial fibroblasts to fas-mediated apoptosis was increased by antisense oligonucleotides targeting the anti-apoptotic gene FLICE-inhibitory protein (FLIP) [46], and increased apoptosis of human RA synovial fibroblasts was induced upon antisense oligonucleotide targeting of galectin-9 [47]. These targets, unsilenced, provide protection against apoptosis, thus maintaining fibroblast populations contributing to persistent inflammation, and as such may be valuable targets in combatting synovial inflammation and hyperplasia. Furthermore, oligonucleotides have been demonstrated to reduce the inflammatory phenotype of activated OA and RA synovial fibroblasts. For example, the inflammatory fibroblast phenotype mediated by leptin, an adipokine found elevated in the synovial fluid of both OA and RA patients [30][48], was inhibited by antisense oligonucleotides targeting the leptin receptor (ObR), which reduced leptin-mediated IL-8 secretion [49] and IL-6 expression in OA fibroblasts [50]. Increasing evidence has emerged that long non-coding RNAs (lncRNAs), such as MALAT1, are central regulators of the inflammatory response [51]. LncRNAs are a relatively novel class of non-coding RNAs, which have been shown to regulate gene expression at both the epigenetic pre-transcriptional and post-transcriptional level through their ability to act as scaffolds for the binding of proteins and other RNAs [52][53][54][55][56]. Recently, the MALAT1 lncRNA was found to regulate the inflammatory response of articular OA chondrocytes [57] and synovial fibroblasts [43]. A locked nucleic acid (LNA) oligonucleotide targeting the MALAT1 lncRNA was found to inhibit both the proliferative and inflammatory phenotype of obese OA synovial fibroblasts [43]. Therefore, oligonucleotide targeting of lncRNAs that are dysregulated in the tissues of the arthritic joint could provide novel therapeutic strategies to target the epigenetic drivers of joint inflammation [55].
As yet, few studies have reported the effect of oligonucleotides designed to target the synovium in preclinical in vivo models of either RA or OA disease [58][59][60][61][62][63]. However, in a surgically induced experimental model of OA, a 21-mer end-capped phosphorothioate antisense directed against Dickkopf-1 (DKK1), the canonical Wnt pathway inhibitor, was intraperitoneally administered at a dose of 20 μg/kg/week for up to 12 weeks in rats and was found to ameliorate synovial vascularity [58]. Similarly, intra-articular injection of FoxC1 siRNA , which is a promoter of TNF-α and IL-8 production in synovial fibroblasts, was found to reduce arthritis development in rodent models of OA and RA [59][60].
3. Oligonucleotides Targeting Subchondral Bone Pathology
In bone homeostasis, receptor activator of nuclear factor kappa B (RANK)/RANKL pathway activates NF-kB-induced transcription factors that provide the balance between bone resorption and bone formation [64][65]. However, this homeostasis is lost in the pro-inflammatory microenvironment within the RA joint, where there is the promotion of bone erosion via the pro-inflammatory cytokine induction of RANKL, which binds to the RANK receptor on osteoclasts and activates their bone resorbing activity [64]. Conversely, in OA the subchondral bone becomes sclerotic, with pronounced trabecular thickening, areas of subchondral bone that are under-mineralised and the formation of bony spurs (osteophytes), which are notable in X-ray radiographs of the joint [30][64][66][67][68]. An often overlooked pathological feature in OA, there is now evidence that subchondral bone changes in OA precede and drive cartilage damage [69][70], with OA bone and OA osteoblasts exhibiting an abnormal type I collagen alpha1 homotrimer phenotype [71] with impaired mineralisation [72]. In human primary osteoblasts, ObR antisense oligonucleotides abolished the leptin-mediated production of oncostatin M [73], which, as a member of the IL6 family, has been associated with bone remodelling and cartilage volume loss in OA and RA [74] as well as ObR itself being associated with biomarkers of cartilage loss and bone remodelling over 2 years in knee OA patients [75]. In vitro, antisense oligonucleotides have also been shown to effectively impact on osteogenic differentiation [76]. In mesenchymal stem cells isolated from patients with knee OA, oligonucleotide antisense against miR29a impaired Wnt-mediated osteogenic differentiation via reducing Wnt3 expression [76].
In vivo, in either a surgically induced OA model anterior cruciate ligament (ACL) transection or a collagenase-induced OA model, end-capped DKK1 antisense oligonucleotides delivered intraperitoneally (10–50 µg/kg/week for up to 8 weeks) lowered disease severity, with a reduction in bone mineral density loss, reduced serum levels of bone resorption markers osteocalcin and CTX-1 and suppressed expression of TNF-α, IL-1β, MMP3 and RANKL [77]. Such findings are consistent with the known role of DKK1 in bone homeostasis as an endogenous inhibitor of the Wnt/beta-catenin signalling pathway. DKK1 is implicated in bone development, the pathological remodelling of bone in both OA and osteoporosis and mediating inflammation-induced bone loss by inhibiting osteoblast differentiation [78][79]. In osteoporosis patients, serum levels of DKK1 are negatively associated with bone mineral density in the femoral head and lumbar spine [80]. More recently, intra-articular injection of an siRNA oligonucleotide targeting YAP, which promotes osteogenesis and bone remodelling, reduced the aberrant subchondral bone formation in the ACL mouse model of OA [81]. Amelioration of sclerotic subchondral bone formation, as well as an overall reduction in OA severity score, has also been achieved in the mouse DMM model by intra-articular delivery of a 2′OME 5′Chol-modified antisense oligonucleotide (2 nmol) targeting the thyroid hormone receptor (THR) [82].
4. Oligonucleotides Targeting Cartilage Degeneration
Cartilage degradation in OA is a key hallmark of OA incidence and of disease progression. This is driven largely by a pathological switch in the phenotype of chondrocytes [30]. In healthy adult cartilage, the chondrocytes are embedded in the extracellular matrix and are in a relatively metabolically inactive state, where they produce type II collagen and aggrecan proteoglycans that hydrate the cartilage and provide the cartilage with its load-absorbing properties [83]. However, in OA the chondrocytes proliferate and become hypertrophic, switching from producing extracellular matrix proteins to producing catabolic MMPs and aggrecanases ADAMTS4 and ADAMTS5, which degrade type II collagen and aggrecan proteoglycan, respectively [30][83]. Induction of MMPs and aggrecanases is promoted by the inflammatory microenvironment of the joint [84], with pro-inflammatory cytokines secreted by various joint tissues and damage-associated molecular patterns (DAMPs) such as fibronectin fragments from degraded cartilage potentiating a vicious cycle of inflammation and cartilage damage via activation of TLRs, the MAPK pathway and the NF-kB pathway [30]. The cellular cross-talk between synovial fibroblasts and chondrocytes is fundamental in this, with IL-6 released from cartilage chondrocytes capable of binding to the soluble IL-6 receptor (sIL-6R) in the synovial fluid and this IL-6/sIL-6R complex transactivating the membrane-bound gp130 on fibroblasts to promote further IL-6 secretion [85]. This chondrocyte–fibroblast crosstalk is further exacerbated in obese patients with OA, where the adipokine leptin stimulates greater IL-6 secretion from articular chondrocytes [85].
In vitro, antisense oligonucleotides have been shown to modulate the inflammatory and catabolic phenotype of human OA chondrocytes. Targeted knockdown of the G-Protein Coupled Receptor (GPCR) RDC1 in human knee OA chondrocytes using antisense oligonucleotides modified the OA chondrocyte phenotype, with reduced expression of a panel of MMPs and hypertrophic markers [86]. Similarly, antisense oligonucleotides targeting either p38 MAPK or the downstream MAPKAPK2 (MK2), which regulates TNF stability via TPP regulation, inhibited IL-1β-induced production of MMP3, MMP13 and PGE2 [29][84]. Modulation of MMP13 expression in human chondrocytes was also reported using c-Fos and c-Jun antisense oligonucleotide, which acted to inhibit the potentiating action of SDF-1alpha on MMP-13 promoter activity [87]. In vivo, intra-articular injection of siRNA antisense targeting MMP13 and ADAMTS5, either alone or in combination, improved histological scores of OA severity in a murine DMM model, compared to non-targeting control siRNA [88].
In recent years, the profiling of non-coding miRNAs has identified a number of miRNAs that are dysregulated in OA diseased cartilage [89], and/or associated with disease progression [90][91], and several studies have now demonstrated modulation of the OA chondrocyte phenotype by antisense oligonucleotide targeting of these miRNAs. In vitro, antisense oligonucleotides against miR-320a reduced the IL-1β-mediated release of MMP13 in human chondrocytes [92], whilst conversely, miRNA oligonucleotide mimics of miR-98 and mir-146 reduced TNF-α and MMP13 production in human OA chondrocytes [89]. In vivo, miR-128-targeting antisense oligonucleotides administered by intra-articular injection were reported to slow articular cartilage degradation, reduce synovitis and slow subchondral bone changes in the ACL experimental model of OA at 8 weeks [93]. In the destabilisation of the medical menisci (DMM) experimental model of OA, miR-181a-5p LNA antisense oligonucleotides delivered by intra-articular injection (3 µL of 1 µg/µL per knee joint) attenuated cartilage destruction [94]. Similarly, intra-articular injection of antagomir-21-5p significantly attenuated the severity of OA in the DMM model, via modulating expression of FGF18 [95]. Furthermore, intra-articular injection of antisense oligonucleotides targeting miR-34a-5p was chondroprotective in a murine DMM model and in a high-fat diet/DMM model [96], whilst LNA antisense inhibition of miR-449a via intra-articular injection (100 nM injections twice weekly for 8 weeks) promoted cartilage regeneration and expression of type II collagen and aggrecan in a rat acute cartilage defect model after 4 and 8 weeks post surgery and in a rat DMM model [97].
In addition to miRNA inhibition, miRNA oligonucleotide mimics have also been demonstrated to have potential as arthritis therapeutics in vivo. A miRNA mimic of miR-26a/26b was found to promote chondrocyte proliferation in vitro by targeting of fucosyltransferase 4 (FUT4) and to attenuate development of OA in a rat ACL model of OA when delivered by single intra-articular injection at a dose of 5 nmol, 1 week after surgical induction of OA [98]. Similarly, intra-articular injection of a miR-145 mimic (at a dose of 50 µM twice a week for 7 weeks) reduced cartilage degradation in a rat DMM model via suppression of MKK4-mediated induction of TNF-α [99]. There is therefore increasing evidence that oligonucleotide therapeutics, when delivered by intra-articular injection, are capable of modulating the diseased inflammatory phenotype of arthritis cartilage.
5. Conclusions
EVs from plasma and SF of OA participants consist of LEVs, MEVs and SEVs that carry cytokines and surface markers related to stem cells and progenitor cells, immune cells, activated pro-inflammatory fibroblasts, epithelial and endothelial cells. Multiple immune cell-derived EV subpopulations were enriched in SF compared with plasma, consistent with OA as an inflammatory arthritis (Figure 5A). The pro-inflammatory phenotype of SF EVs was supported by their pro-inflammatory cytokine cargo (Figure 5A). In contrast, HSC-, progenitor cell-, and endothelial cell-associated EV populations were enriched in plasma relative to SF (Figure 5B). Ratios of neutrophil-EVs to lymphocyte-EVs were positively correlated between plasma and SF (Figure 5C); the ability to derive ratios of neutrophils to lymphocytes from frozen samples by EV profiling can potentially provide a powerful biomarker of OA pathology and other comorbidities, such as cardiovascular disease. EVs related to several types of stem cells, progenitor cells, neutrophils and B cells, and endo-EV pro-inflammatory cytokines IL-6 and TNF-α were highly correlated between SF and plasma (Figure 5D), suggesting plasma EVs have the potential to reflect OA joint inflammation and disease severity. These subpopulations in particular may be direct biomarkers of disease, involved in disease pathogenesis and informing on disease activity.

References
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15.
- Crowson, C.S.; Matteson, E.L.; Myasoedova, E.; Michet, C.J.; Ernste, F.C.; Warrington, K.J.; Davis, J.M.; Hunder, G.G.; Therneau, T.M.; Gabriel, S.E. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum. 2011, 63, 633–639.
- Crane, M.M.; Juneja, M.; Allen, J.; Kurrasch, R.H.; Chu, M.E.; Quattrocchi, E.; Manson, S.C.; Chang, D.J. Epidemiology and Treatment of New-Onset and Established Rheumatoid Arthritis in an Insured US Population. Arthritis Care Res. 2015, 67, 1646–1655.
- Lawrence, R.C.; Felson, D.T.; Helmick, C.G.; Arnold, L.M.; Choi, H.; Deyo, R.A.; Gabriel, S.; Hirsch, R.; Hochberg, M.C.; Hunder, G.G.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008, 58, 26–35.
- Chaudhari, K.; Rizvi, S.; Syed, B.A. Rheumatoid arthritis: Current and future trends. Nat. Rev. Drug Discov. 2016, 15, 305–306.
- Hua, C.; Buttgereit, F.; Combe, B. Glucocorticoids in rheumatoid arthritis: Current status and future studies. RMD Open 2020, 6.
- Briot, K.; Roux, C. Glucocorticoid-induced osteoporosis. RMD Open 2015, 1, e000014.
- Schakman, O.; Kalista, S.; Barbe, C.; Loumaye, A.; Thissen, J.P. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172.
- van Vollenhoven, R. Treat-to-target in rheumatoid arthritis—Are we there yet? Nat. Rev. Rheumatol. 2019, 15, 180–186.
- Pokharel, G.; Deardon, R.; Johnson, S.R.; Tomlinson, G.; Hull, P.M.; Hazlewood, G.S. Effectiveness of initial methotrexate-based treatment approaches in early rheumatoid arthritis: An elicitation of rheumatologists’ beliefs. Rheumatology 2020.
- Friedman, B.; Cronstein, B. Methotrexate mechanism in treatment of rheumatoid arthritis. Jt. Bone Spine 2019, 86, 301–307.
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat. Rev. Rheumatol. 2016, 12, 731–742.
- Fox, R.I.; Herrmann, M.L.; Frangou, C.G.; Wahl, G.M.; Morris, R.E.; Strand, V.; Kirschbaum, B.J. Mechanism of action for leflunomide in rheumatoid arthritis. Clin. Immunol. 1999, 93, 198–208.
- Hua, L.; Du, H.; Ying, M.; Wu, H.; Fan, J.; Shi, X. Efficacy and safety of low-dose glucocorticoids combined with methotrexate and hydroxychloroquine in the treatment of early rheumatoid arthritis: A single-center, randomized, double-blind clinical trial. Medicine 2020, 99, e20824.
- Schrezenmeier, E.; Dorner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166.
- Smedegard, G.; Bjork, J. Sulphasalazine: Mechanism of action in rheumatoid arthritis. Br. J. Rheumatol. 1995, 34 (Suppl. 2), 7–15.
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: A potent and specific inhibitor of nuclear factor kappa B. J. Clin. Investig. 1998, 101, 1163–1174.
- Edrees, A.F.; Misra, S.N.; Abdou, N.I. Anti-tumor necrosis factor (TNF) therapy in rheumatoid arthritis: Correlation of TNF-alpha serum level with clinical response and benefit from changing dose or frequency of infliximab infusions. Clin. Exp. Rheumatol. 2005, 23, 469–474.
- Wu, C.; Wang, S.; Xian, P.; Yang, L.; Chen, Y.; Mo, X. Effect of Anti-TNF Antibodies on Clinical Response in Rheumatoid Arthritis Patients: A Meta-Analysis. Biomed. Res. Int. 2016, 2016, 7185708.
- Mathieu, S.; Couderc, M.; Pereira, B.; Soubrier, M. The effects of TNF-alpha inhibitor therapy on arterial stiffness and endothelial dysfunction in rheumatoid arthritis: A meta-analysis. Semin. Arthritis Rheum. 2013, 43, e1–e2.
- Ma, X.; Xu, S. TNF inhibitor therapy for rheumatoid arthritis. Biomed. Rep. 2013, 1, 177–184.
- Biggioggero, M.; Crotti, C.; Becciolini, A.; Favalli, E.G. Tocilizumab in the treatment of rheumatoid arthritis: An evidence-based review and patient selection. Drug Des. Devel. Ther. 2019, 13, 57–70.
- Cohen, M.D.; Keystone, E. Rituximab for Rheumatoid Arthritis. Rheumatol. Ther. 2015, 2, 99–111.
- Vital, E.M.; Emery, P. Abatacept in the treatment of rheumatoid arthritis. Ther. Clin. Risk Manag. 2006, 2, 365–375.
- Wang, W.; Zhou, H.; Liu, L. Side effects of methotrexate therapy for rheumatoid arthritis: A systematic review. Eur. J. Med. Chem. 2018, 158, 502–516.
- Aletaha, D.; Kapral, T.; Smolen, J.S. Toxicity profiles of traditional disease modifying antirheumatic drugs for rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 482–486.
- Moots, R.J.; Xavier, R.M.; Mok, C.C.; Rahman, M.U.; Tsai, W.C.; Al-Maini, M.H.; Pavelka, K.; Mahgoub, E.; Kotak, S.; Korth-Bradley, J.; et al. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with adalimumab, etanercept, or infliximab: Results from a multinational, real-world clinical practice, non-interventional study. PLoS ONE 2017, 12, e0175207.
- Safiri, S.; Kolahi, A.A.; Smith, E.; Hill, C.; Bettampadi, D.; Mansournia, M.A.; Hoy, D.; Ashrafi-Asgarabad, A.; Sepidarkish, M.; Almasi-Hashiani, A.; et al. Global, regional and national burden of osteoarthritis 1990–2017: A systematic analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis. 2020, 79, 819–828.
- Philp, A.M.; Davis, E.T.; Jones, S.W. Developing anti-inflammatory therapeutics for patients with osteoarthritis. Rheumatology 2017, 56, 869–881.
- Tonge, D.P.; Pearson, M.J.; Jones, S.W. The hallmarks of osteoarthritis and the potential to develop personalised disease-modifying pharmacological therapeutics. Osteoarthr. Cartil. 2014, 22, 609–621.
- Gato-Calvo, L.; Magalhaes, J.; Ruiz-Romero, C.; Blanco, F.J.; Burguera, E.F. Platelet-rich plasma in osteoarthritis treatment: Review of current evidence. Ther. Adv. Chronic Dis. 2019, 10, 2040622319825567.
- Ogata, T.; Ideno, Y.; Akai, M.; Seichi, A.; Hagino, H.; Iwaya, T.; Doi, T.; Yamada, K.; Chen, A.Z.; Li, Y.; et al. Effects of glucosamine in patients with osteoarthritis of the knee: A systematic review and meta-analysis. Clin. Rheumatol. 2018, 37, 2479–2487.
- Maricar, N.; Parkes, M.J.; Callaghan, M.J.; Hutchinson, C.E.; Gait, A.D.; Hodgson, R.; Felson, D.T.; O’Neill, T.W. Structural predictors of response to intra-articular steroid injection in symptomatic knee osteoarthritis. Arthritis Res. Ther. 2017, 19, 88.
- Evans, C.H. Drug delivery to chondrocytes. Osteoarthr. Cartil. 2016, 24, 1–3.
- Ospelt, C. Synovial fibroblasts in 2017. RMD Open 2017, 3, e000471.
- Farah, H.; Young, S.P.; Mauro, C.; Jones, S.W. Metabolic dysfunction and inflammatory disease: The role of stromal fibroblasts. FEBS J. 2020.
- Kung, C.C.; Dai, S.P.; Chiang, H.; Huang, H.S.; Sun, W.H. Temporal expression patterns of distinct cytokines and M1/M2 macrophage polarization regulate rheumatoid arthritis progression. Mol. Biol. Rep. 2020, 47, 3423–3437.
- Ostergaard, M.; Peterfy, C.; Conaghan, P.; McQueen, F.; Bird, P.; Ejbjerg, B.; Shnier, R.; O’Connor, P.; Klarlund, M.; Emery, P.; et al. OMERACT Rheumatoid Arthritis Magnetic Resonance Imaging Studies. Core set of MRI acquisitions, joint pathology definitions, and the OMERACT RA-MRI scoring system. J. Rheumatol. 2003, 30, 1385–1386.
- Myers, S.L.; Brandt, K.D.; Ehlich, J.W.; Braunstein, E.M.; Shelbourne, K.D.; Heck, D.A.; Kalasinski, L.A. Synovial inflammation in patients with early osteoarthritis of the knee. J. Rheumatol. 1990, 17, 1662–1669.
- Rhodes, L.A.; Conaghan, P.G.; Radjenovic, A.; Grainger, A.J.; Emery, P.; McGonagle, D. Further evidence that a cartilage-pannus junction synovitis predilection is not a specific feature of rheumatoid arthritis. Ann. Rheum. Dis. 2005, 64, 1347–1349.
- Fernandez-Madrid, F.; Karvonen, R.L.; Teitge, R.A.; Miller, P.R.; An, T.; Negendank, W.G. Synovial thickening detected by MR imaging in osteoarthritis of the knee confirmed by biopsy as synovitis. Magn. Reson. Imaging 1995, 13, 177–183.
- Oehler, S.; Neureiter, D.; Meyer-Scholten, C.; Aigner, T. Subtyping of osteoarthritic synoviopathy. Clin. Exp. Rheumatol. 2002, 20, 633–640.
- Nanus, D.E.; Wijesinghe, S.N.; Pearson, M.J.; Hadjicharalambous, M.R.; Rosser, A.; Davis, E.T.; Lindsay, M.A.; Jones, S.W. Regulation of the Inflammatory Synovial Fibroblast Phenotype by Metastasis-Associated Lung Adenocarcinoma Transcript 1 Long Noncoding RNA in Obese Patients With Osteoarthritis. Arthritis Rheumatol. 2020, 72, 609–619.
- Nakazawa, M.; Ishii, H.; Aono, H.; Takai, M.; Honda, T.; Aratani, S.; Fukamizu, A.; Nakamura, H.; Yoshino, S.; Kobata, T.; et al. Role of Notch-1 intracellular domain in activation of rheumatoid synoviocytes. Arthritis Rheum. 2001, 44, 1545–1554.
- Stanford, S.M.; Maestre, M.F.; Campbell, A.M.; Bartok, B.; Kiosses, W.B.; Boyle, D.L.; Arnett, H.A.; Mustelin, T.; Firestein, G.S.; Bottini, N. Protein tyrosine phosphatase expression profile of rheumatoid arthritis fibroblast-like synoviocytes: A novel role of SH2 domain-containing phosphatase 2 as a modulator of invasion and survival. Arthritis Rheum. 2013, 65, 1171–1180.
- Palao, G.; Santiago, B.; Galindo, M.; Paya, M.; Ramirez, J.C.; Pablos, J.L. Down-regulation of FLIP sensitizes rheumatoid synovial fibroblasts to Fas-mediated apoptosis. Arthritis Rheum. 2004, 50, 2803–2810.
- Pearson, M.J.; Bik, M.A.; Ospelt, C.; Naylor, A.J.; Wehmeyer, C.; Jones, S.W.; Buckley, C.D.; Gay, S.; Filer, A.; Lord, J.M. Endogenous Galectin-9 Suppresses Apoptosis in Human Rheumatoid Arthritis Synovial Fibroblasts. Sci. Rep. 2018, 8, 12887.
- Toussirot, E.; Streit, G.; Wendling, D. The contribution of adipose tissue and adipokines to inflammation in joint diseases. Curr. Med. Chem. 2007, 14, 1095–1100.
- Tong, K.M.; Shieh, D.C.; Chen, C.P.; Tzeng, C.Y.; Wang, S.P.; Huang, K.C.; Chiu, Y.C.; Fong, Y.C.; Tang, C.H. Leptin induces IL-8 expression via leptin receptor, IRS-1, PI3K, Akt cascade and promotion of NF-kappaB/p300 binding in human synovial fibroblasts. Cell Signal. 2008, 20, 1478–1488.
- Yang, W.H.; Liu, S.C.; Tsai, C.H.; Fong, Y.C.; Wang, S.J.; Chang, Y.S.; Tang, C.H. Leptin induces IL-6 expression through OBRl receptor signaling pathway in human synovial fibroblasts. PLoS ONE 2013, 8, e75551.
- Huang, M.; Wang, H.; Hu, X.; Cao, X. lncRNA MALAT1 binds chromatin remodeling subunit BRG1 to epigenetically promote inflammation-related hepatocellular carcinoma progression. Oncoimmunology 2019, 8, e1518628.
- Hamann, P.D.; Roux, B.T.; Heward, J.A.; Love, S.; McHugh, N.J.; Jones, S.W.; Lindsay, M.A. Transcriptional profiling identifies differential expression of long non-coding RNAs in Jo-1 associated and inclusion body myositis. Sci. Rep. 2017, 7, 8024.
- Heward, J.A.; Lindsay, M.A. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 2014, 35, 408–419.
- Ilott, N.E.; Heward, J.A.; Roux, B.; Tsitsiou, E.; Fenwick, P.S.; Lenzi, L.; Goodhead, I.; Hertz-Fowler, C.; Heger, A.; Hall, N.; et al. Corrigendum: Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat. Commun. 2015, 6, 6814.
- Pearson, M.J.; Jones, S.W. Review: Long Noncoding RNAs in the Regulation of Inflammatory Pathways in Rheumatoid Arthritis and Osteoarthritis. Arthritis Rheumatol. 2016, 68, 2575–2583.
- Roux, B.T.; Heward, J.A.; Donnelly, L.E.; Jones, S.W.; Lindsay, M.A. Catalog of Differentially Expressed Long Non-Coding RNA following Activation of Human and Mouse Innate Immune Response. Front. Immunol. 2017, 8, 1038.
- Pearson, M.J.; Philp, A.M.; Heward, J.A.; Roux, B.T.; Walsh, D.A.; Davis, E.T.; Lindsay, M.A.; Jones, S.W. Long Intergenic Noncoding RNAs Mediate the Human Chondrocyte Inflammatory Response and Are Differentially Expressed in Osteoarthritis Cartilage. Arthritis Rheumatol. 2016, 68, 845–856.
- Weng, L.H.; Ko, J.Y.; Wang, C.J.; Sun, Y.C.; Wang, F.S. Dkk-1 promotes angiogenic responses and cartilage matrix proteinase secretion in synovial fibroblasts from osteoarthritic joints. Arthritis Rheum. 2012, 64, 3267–3277.
- Wang, J.; Wang, Y.; Zhang, H.; Gao, W.; Lu, M.; Liu, W.; Li, Y.; Yin, Z. Forkhead box C1 promotes the pathology of osteoarthritis by upregulating beta-catenin in synovial fibroblasts. FEBS J. 2020, 287, 3065–3087.
- Wang, J.; Wang, Y.; Zhang, H.; Chang, J.; Lu, M.; Gao, W.; Liu, W.; Li, Y.; Yin, L.; Wang, X.; et al. Identification of a novel microRNA-141-3p/Forkhead box C1/β-catenin axis associated with rheumatoid arthritis synovial fibroblast function in vivo and in vitro. Theranostics 2020, 10, 5412–5434.
- Jia, W.; Wu, W.; Yang, D.; Xiao, C.; Huang, M.; Long, F.; Su, Z.; Qin, M.; Liu, X.; Zhu, Y.Z. GATA4 regulates angiogenesis and persistence of inflammation in rheumatoid arthritis. Cell Death Dis. 2018, 9, 503.
- Li, D.; Xiao, Z.; Wang, G.; Song, X. Knockdown of ADAM10 inhibits migration and invasion of fibroblast-like synoviocytes in rheumatoid arthritis. Mol. Med. Rep. 2015, 12, 5517–5523.
- Wang, H.; Chen, W.; Wang, L.; Li, F.; Zhang, C.; Xu, L. Tumor necrosis factor receptor-associated factor 6 promotes migration of rheumatoid arthritis fibroblast-like synoviocytes. Mol. Med. Rep. 2015, 11, 2761–2766.
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673.
- Kim, J.H.; Kim, N. Signaling Pathways in Osteoclast Differentiation. Chonnam. Med. J. 2016, 52, 12–17.
- Hunter, D.J.; Gerstenfeld, L.; Bishop, G.; Davis, A.D.; Mason, Z.D.; Einhorn, T.A.; Maciewicz, R.A.; Newham, P.; Foster, M.; Jackson, S.; et al. Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res. Ther. 2009, 11, R11.
- Bailey, A.J. Changes in bone collagen with age and disease. J. Musculoskelet Neuronal Interact. 2002, 2, 529–531.
- Bailey, A.J.; Sims, T.J.; Knott, L. Phenotypic expression of osteoblast collagen in osteoarthritic bone: Production of type I homotrimer. Int. J. Biochem. Cell Biol. 2002, 34, 176–182.
- Quasnichka, H.L.; Anderson-MacKenzie, J.M.; Bailey, A.J. Subchondral bone and ligament changes precede cartilage degradation in guinea pig osteoarthritis. Biorheology 2006, 43, 389–397.
- Radin, E.L.; Rose, R.M. Role of subchondral bone in the initiation and progression of cartilage damage. Clin. Orthop. Relat. Res. 1986, 213, 34–40.
- Philp, A.M.; Collier, R.L.; Grover, L.M.; Davis, E.T.; Jones, S.W. Resistin promotes the abnormal Type I collagen phenotype of subchondral bone in obese patients with end stage hip osteoarthritis. Sci. Rep. 2017, 7, 4042.
- Chang, J.; Jackson, S.G.; Wardale, J.; Jones, S.W. Hypoxia modulates the phenotype of osteoblasts isolated from knee osteoarthritis patients, leading to undermineralized bone nodule formation. Arthritis Rheumatol. 2014, 66, 1789–1799.
- Yang, W.H.; Tsai, C.H.; Fong, Y.C.; Huang, Y.L.; Wang, S.J.; Chang, Y.S.; Tang, C.H. Leptin induces oncostatin M production in osteoblasts by downregulating miR-93 through the Akt signaling pathway. Int. J. Mol. Sci. 2014, 15, 15778–15790.
- Lisignoli, G.; Toneguzzi, S.; Pozzi, C.; Piacentini, A.; Riccio, M.; Ferruzzi, A.; Gualtieri, G.; Facchini, A. Proinflammatory cytokines and chemokine production and expression by human osteoblasts isolated from patients with rheumatoid arthritis and osteoarthritis. J. Rheumatol. 1999, 26, 791–799.
- Berry, P.A.; Jones, S.W.; Cicuttini, F.M.; Wluka, A.E.; Maciewicz, R.A. Temporal relationship between serum adipokines, biomarkers of bone and cartilage turnover, and cartilage volume loss in a population with clinical knee osteoarthritis. Arthritis Rheum. 2011, 63, 700–707.
- Lian, W.S.; Wu, R.W.; Lee, M.S.; Chen, Y.S.; Sun, Y.C.; Wu, S.L.; Ke, H.J.; Ko, J.Y.; Wang, F.S. Subchondral mesenchymal stem cells from osteoarthritic knees display high osteogenic differentiation capacity through microRNA-29a regulation of HDAC4. J. Mol. Med. 2017, 95, 1327–1340.
- Weng, L.H.; Wang, C.J.; Ko, J.Y.; Sun, Y.C.; Wang, F.S. Control of Dkk-1 ameliorates chondrocyte apoptosis, cartilage destruction, and subchondral bone deterioration in osteoarthritic knees. Arthritis Rheum. 2010, 62, 1393–1402.
- Qiang, Y.W.; Barlogie, B.; Rudikoff, S.; Shaughnessy, J.D., Jr. Dkk1-induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone 2008, 42, 669–680.
- Hurson, C.J.; Butler, J.S.; Keating, D.T.; Murray, D.W.; Sadlier, D.M.; O’Byrne, J.M.; Doran, P.P. Gene expression analysis in human osteoblasts exposed to dexamethasone identifies altered developmental pathways as putative drivers of osteoporosis. BMC Musculoskelet Disord. 2007, 8, 12.
- Butler, J.S.; Murray, D.W.; Hurson, C.J.; O’Brien, J.; Doran, P.P.; O’Byrne, J.M. The role of Dkk1 in bone mass regulation: Correlating serum Dkk1 expression with bone mineral density. J. Orthop. Res. 2011, 29, 414–418.
- Gong, Y.; Li, S.J.; Liu, R.; Zhan, J.F.; Tan, C.; Fang, Y.F.; Chen, Y.; Yu, B. Inhibition of YAP with siRNA prevents cartilage degradation and ameliorates osteoarthritis development. J. Mol. Med. 2019, 97, 103–114.
- Li, L.; Li, M.; Pang, Y.; Wang, J.; Wan, Y.; Zhu, C.; Yin, Z. Abnormal thyroid hormone receptor signaling in osteoarthritic osteoblasts regulates microangiogenesis in subchondral bone. Life Sci. 2019, 239, 116975.
- Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177–192.
- Jones, S.W.; Brockbank, S.M.; Clements, K.M.; Le Good, N.; Campbell, D.; Read, S.J.; Needham, M.R.; Newham, P. Mitogen-activated protein kinase-activated protein kinase 2 (MK2) modulates key biological pathways associated with OA disease pathology. Osteoarthr. Cartil. 2009, 17, 124–131.
- Pearson, M.J.; Herndler-Brandstetter, D.; Tariq, M.A.; Nicholson, T.A.; Philp, A.M.; Smith, H.L.; Davis, E.T.; Jones, S.W.; Lord, J.M. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci. Rep. 2017, 7, 3451.
- Jones, S.W.; Brockbank, S.M.; Mobbs, M.L.; Le Good, N.J.; Soma-Haddrick, S.; Heuze, A.J.; Langham, C.J.; Timms, D.; Newham, P.; Needham, M.R. The orphan G-protein coupled receptor RDC1: Evidence for a role in chondrocyte hypertrophy and articular cartilage matrix turnover. Osteoarthr. Cartil. 2006, 14, 597–608.
- Chiu, Y.C.; Yang, R.S.; Hsieh, K.H.; Fong, Y.C.; Way, T.D.; Lee, T.S.; Wu, H.C.; Fu, W.M.; Tang, C.H. Stromal cell-derived factor-1 induces matrix metalloprotease-13 expression in human chondrocytes. Mol. Pharmacol. 2007, 72, 695–703.
- Hoshi, H.; Akagi, R.; Yamaguchi, S.; Muramatsu, Y.; Akatsu, Y.; Yamamoto, Y.; Sasaki, T.; Takahashi, K.; Sasho, T. Effect of inhibiting MMP13 and ADAMTS5 by intra-articular injection of small interfering RNA in a surgically induced osteoarthritis model of mice. Cell Tissue Res. 2017, 368, 379–387.
- Jones, S.W.; Watkins, G.; Le Good, N.; Roberts, S.; Murphy, C.L.; Brockbank, S.M.; Needham, M.R.; Read, S.J.; Newham, P. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-alpha and MMP13. Osteoarthr. Cartil. 2009, 17, 464–472.
- Goldring, M.B.; Marcu, K.B. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol. Med. 2012, 18, 109–118.
- Kung, L.H.W.; Ravi, V.; Rowley, L.; Angelucci, C.; Fosang, A.J.; Bell, K.M.; Little, C.B.; Bateman, J.F. Cartilage MicroRNA Dysregulation During the Onset and Progression of Mouse Osteoarthritis Is Independent of Aggrecanolysis and Overlaps With Candidates From End-Stage Human Disease. Arthritis Rheumatol. 2018, 70, 383–395.
- Jin, Y.; Chen, X.; Gao, Z.Y.; Liu, K.; Hou, Y.; Zheng, J. The role of miR-320a and IL-1beta in human chondrocyte degradation. Bone Jt. Res. 2017, 6, 196–203.
- Lian, W.S.; Ko, J.Y.; Wu, R.W.; Sun, Y.C.; Chen, Y.S.; Wu, S.L.; Weng, L.H.; Jahr, H.; Wang, F.S. MicroRNA-128a represses chondrocyte autophagy and exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis. 2018, 9, 919.
- Nakamura, A.; Rampersaud, Y.R.; Nakamura, S.; Sharma, A.; Zeng, F.; Rossomacha, E.; Ali, S.A.; Krawetz, R.; Haroon, N.; Perruccio, A.V.; et al. microRNA-181a-5p antisense oligonucleotides attenuate osteoarthritis in facet and knee joints. Ann. Rheum. Dis. 2019, 78, 111–121.
- Wang, X.B.; Zhao, F.C.; Yi, L.H.; Tang, J.L.; Zhu, Z.Y.; Pang, Y.; Chen, Y.S.; Li, D.Y.; Guo, K.J.; Zheng, X. MicroRNA-21-5p as a novel therapeutic target for osteoarthritis. Rheumatology 2019.
- Endisha, H.; Datta, P.; Sharma, A.; Nakamura, S.; Rossomacha, E.; Younan, C.; Ali, S.A.; Tavallaee, G.; Lively, S.; Potla, P.; et al. MicroRNA-34a-5p Promotes Joint Destruction During Osteoarthritis. Arthritis Rheumatol. 2021, 73, 426–439.
- Baek, D.; Lee, K.M.; Park, K.W.; Suh, J.W.; Choi, S.M.; Park, K.H.; Lee, J.W.; Kim, S.H. Inhibition of miR-449a Promotes Cartilage Regeneration and Prevents Progression of Osteoarthritis in In Vivo Rat Models. Mol. Ther. Nucleic Acids 2018, 13, 322–333.
- Hu, J.; Wang, Z.; Pan, Y.; Ma, J.; Miao, X.; Qi, X.; Zhou, H.; Jia, L. MiR-26a and miR-26b mediate osteoarthritis progression by targeting FUT4 via NF-kappaB signaling pathway. Int. J. Biochem. Cell Biol. 2018, 94, 79–88.
- Hu, G.; Zhao, X.; Wang, C.; Geng, Y.; Zhao, J.; Xu, J.; Zuo, B.; Zhao, C.; Wang, C.; Zhang, X. MicroRNA-145 attenuates TNF-alpha-driven cartilage matrix degradation in osteoarthritis via direct suppression of MKK4. Cell Death Dis. 2017, 8, e3140.


