Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alice Prince | + 2032 word(s) | 2032 | 2021-08-03 04:08:07 | | | |
| 2 | Bruce Ren | -21 word(s) | 2011 | 2021-08-06 07:36:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Prince, A. Pseudomonas aeruginosa Promotes Lung Infection. Encyclopedia. Available online: https://encyclopedia.pub/entry/12844 (accessed on 25 July 2026).
Prince A. Pseudomonas aeruginosa Promotes Lung Infection. Encyclopedia. Available at: https://encyclopedia.pub/entry/12844. Accessed July 25, 2026.
Prince, Alice. "Pseudomonas aeruginosa Promotes Lung Infection" Encyclopedia, https://encyclopedia.pub/entry/12844 (accessed July 25, 2026).
Prince, A. (2021, August 05). Pseudomonas aeruginosa Promotes Lung Infection. In Encyclopedia. https://encyclopedia.pub/entry/12844
Prince, Alice. "Pseudomonas aeruginosa Promotes Lung Infection." Encyclopedia. Web. 05 August, 2021.
Copy Citation
Prevailing dogma indicates that the lung of cystic fibrosis (CF) individuals is infected by multiple pathogens due to the abundant accumulation of mucus, which traps most of inhaled organisms. However, this hypothesis does not explain how specific opportunists, like Pseudomonas aeruginosa, are selected in the CF lung to cause chronic disease. This strongly suggests that other factors than mucus are accrued in the human airway and might predispose to bacterial disease, especially by P. aeruginosa. In this review we discuss the role of macrophage metabolites, like succinate and itaconate, in P. aeruginosa pneumonia.
Pseudomonas aeruginosa
immunometabolism
succinate
itaconate
PTEN
CFTR
cystic fibrosis
inflammation
1. Introduction
Pseudomonas aeruginosa predominates as a major cause of lung infection and pulmonary pathology in patients with cystic fibrosis [1][2]. While other Gram-negative, often antibiotic resistant organisms, also infect these patients, primarily late in the course of lung disease, P. aeruginosa can occur at any stage in cystic fibrosis (CF), but most typically superinfects, and then replaces Staphylococcus aureus as the major airway pathogen [3][4]. Impaired mucociliary clearance and dehydrated airway surface fluid is likely to impact overall bacterial clearance in the CF transmembrane conductance regulator (CFTR)-mutant lung [5], but in itself does not explain why P. aeruginosa, but rarely Klebsiellae, Escherichia coli, Proteus or the other common opportunists, is so specific for CF. Metabolomic data derived from human and murine airways suggest that specific airway metabolites and especially reactive oxygen species (ROS) in general, drive the selection of the specific P. aeruginosa phenotypes that are associated with intractable infection [6].
Opportunistic pathogens, as the name implies, take advantage of local conditions and can adjust gene expression accordingly. This occurs through genetic adaptation, the up or down regulation of specific pathways based upon the environment [6][7]. Bacterial communities respond to secreted molecules involved in transcriptional activation, often through quorum sensing [8]. There is also in vivo selection of mutants, through single nucleotide polymorphisms (SNPs) or the uptake of foreign genes that have favorable characteristics and provide a competitive advantage [9][10]. By studying gene expression in clinical isolates of P. aeruginosa from CF patients with established infection, it is possible to follow the in vivo evolution of specific bacterial genes that are important in chronic infection.
2. The Generation of Succinate in the Airway Provides a Preferred Substrate for P. aeruginosa Proliferation
Historically, bacteria have been classified by their metabolic activity which roughly correlates with the tissues that are major sites of infection [11]. Directed by small RNAs and the crc locus, P. aeruginosa preferentially consumes succinate over other carbon sources until it is depleted, through a process named catabolite repression [12][13][14][15][16]. Thus, in a setting replete with succinate, such as the inflamed airways, P. aeruginosa would have a supply of a major carbon source. Of note, succinate is one of the major metabolites released by macrophages activated by lipopolysaccharide (LPS) [17][18]. Activated macrophages undergo metabolic reprogramming, switching to generate ATP by aerobic glycolysis instead of oxidative phosphorylation (OXPHOS) pathways in the mitochondrion [19]. This metabolic switch repurposes succinate oxidation in the mitochondria not to produce ATP, but, instead, to release bactericidal ROS by action of succinate dehydrogenase (SDH) (complex II) and isocitrate dehydrogenase (IDH, complex I) [19] (Figure 1A). Succinate influx into the active site of SDH is potentiated by anerplerosis, a biochemical process that favors both synthesis and accumulation of succinate from foreign metabolites, such as environmental glutamine [18][19]. Thus, by activating an inflammatory response through LPS, P. aeruginosa provides itself with a favored substrate, succinate (Figure 1A). Even in the normal lung, activated macrophages produce and oxidize succinate, which stimulates both stabilization of the hypoxia-induced factor 1α (HIF-1α) and generation of the potent proinflammatory cytokine IL-1β [17][18][19][20]. As described below, a greater production of succinate in cells with CFTR dysfunction may favor P. aeruginosa proliferation [21] (Figure 1B). Thus, the CFTR dysfunction and excess proinflammatory signaling fuels P. aeruginosa growth, possibly to a greater extent than the recruited immune cells can clear the organisms.
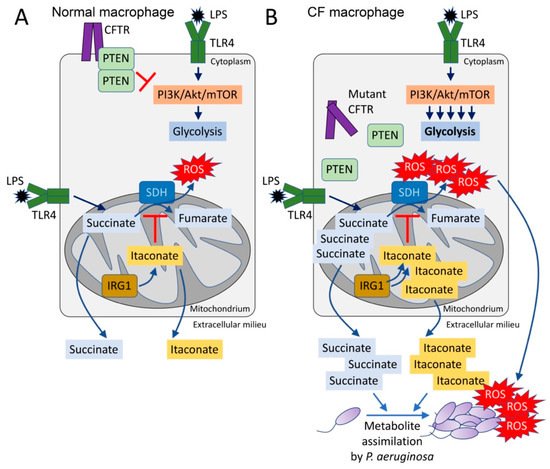
Figure 1. The CFTR-PTEN complex regulates macrophage metabolism and release of immunometabolites. (A) During LPS recognition by TLR4, macrophages activate PI3K-Akt-mTOR signaling and glycolysis. The CFTR-PTEN complex regulates this process by inhibiting PI3K function. In parallel, TLR4-LPS interaction promotes anerplerosis, which replenishes the host mitochondria with succinate. Succinate is oxidized by succinate dehydrogenase (SDH) to fumarate, which produces bactericidal ROS. Pro-oxidant SDH activity is regulated by IRG1, which produces itaconate that competes with succinate for the active site of SDH. (B) In the absence of the CFTR-PTEN complex, glycolysis is overactivated, as well as succinate oxidation. Itaconate is overproduced to compensate for succinate oxidation, leading to both succinate and itaconate accumulation in the mitochondria. Both metabolites are abundantly released out of the cell, where they can be assimilated by Pseudomonas aeruginosa. These organisms also sense ROS, which promotes adaptive changes like biofilm formation.
3. Excess Succinate Release Is a Consequence of CFTR-PTEN Complex Dysfunction
Increased succinate release is a property of CF cells, even in the absence of infection [21]. Cellular metabolic activity is controlled by PI3K and its phosphatase PTEN, an interaction which regulates phosphoinositide abundance, downstream Akt/mTOR signaling and ultimately TCA cycle function in the mitochondrion [22][23][24][25][26]. Functional PTEN is associated with CFTR at the cell membrane, enabling its dimerization and de-phosphorylation [21] (Figure 1A). In the absence of sufficient membrane bound CFTR, PTEN activity is impaired and its brake on mitochondrial generation of succinate is released [21][25]. Mechanistically, lack of membrane PTEN in CF cells favors increased glycolysis and repurposing of mitochondria to produce ROS instead of ATP (Figure 1B). Accumulation of pro-oxidant species activates a compensatory mechanism led by the synthesis of itaconate (cis-itaconate), another TCA cycle intermediate produced by Irg1 (immunoregulatory gene 1, also known as Acod1) that inhibits SDH function [21][27] (Figure 1B). Thus, excessive succinate oxidation is prevented by itaconate, but succinate accumulates and permeates towards extracellular compartments where P. aeruginosa both senses and assimilates it as carbon source [13][21][28] (Figure 1B). The release of excess succinate by cells with Cftr mutations can be corrected by restoring sufficient amounts of PTEN [21]. This also reduces ROS production by mitochondria. Increasing the delivery of CFTR to the membrane would then provide docking sites for PTEN which would also serve to normalize succinate, a response that might be associated with the highly active CFTR modulator therapy, although this has not been directly examined.
The poor PTEN-CFTR interaction associated with increased succinate in CF is also involved in the altered NF-κB signaling in the airway. PTEN regulates the immunostimulatory functions of the Akt/mTOR pathway [25]. In the absence of sufficient PTEN, the Toll-like receptor 4 (TLR4) adaptor TIRAP/MAL is decreased along with the immunoregulatory p110δ component of PI3K [25]. This results in increased proinflammatory cytokine production during P. aeruginosa pneumonia, and may explain the observation that CF infants and some animal models (e.g., ferret) have elevated proinflammatory cytokines in otherwise seemingly normal airways [29][30][31].
4. Succinate and the Production of Reactive Oxygen Species in the CF Airway
Succinate, a major component of the TCA cycle, is produced as a function of both bacterial and host metabolism through metabolic pathways that generate oxidants. The specific metabolic pathways used by both host and pathogen to generate ATP produce ROS to differing amounts. While ROS generated intracellularly by phagocytes is important in bacterial killing [32], specially from phagosomes and by complex I and II in mitochondrial compartments, oxidant species are major byproducts of metabolic activity with potentially detrimental effects for both the host airway and the pathogen. Several clinical studies in CF have correlated markers of oxidants stress, such as isoprostane, with inflammation and decreased pulmonary function [33]. As discussed above, lack of normal CFTR function generates excess ROS in many cell types in the airway, independent of infection [34]. ROS inhibits autophagy in CF cells, inducing aggresome formation which adds to inflammation [34]. When sensed by bacteria, excess ROS causes protein aggregation and evokes a major bacterial anti-oxidant response [35]. CF respiratory pathogens have been noted to have significantly increased anti-oxidant capacity as compared with commensal flora or even other respiratory pathogens [36], which is consistent with selection under the increased oxidant stress found in the CF airway. Of note, numerous clinical studies have attempted to therapeutically decrease airway oxidants in CF with a variety of drugs, but without substantial success [37], potentially due to the presence of already ROS-adapted strains that persist beyond the type of treatment used.
The presence of succinate in the CF airway provides a milieu that supports P. aeruginosa proliferation with a preferred substrate. Bacteria grown under conditions of high succinate, like those found in the CF airway, activated pyroptosis and macrophage death, and generated greater amounts of the potent cytokine IL-1β and more succinate [21]. These succinate-adapted strains were better able to colonize the murine airways. However, the endogenous oxidant stress fueled by succinate and generated by Pseudomonas metabolic activity as well provides selective pressure for adaptive changes [21]. In response to high succinate in both LB and artificial sputum media (ASM), P. aeruginosa PAO1 diverts glucose metabolism via the glyoxylate shunt and Entner-Doudoroff pathway to produce extracellular polysaccharides (EPSs), like alginate, and biofilm [6][21]. These pathways generate fewer oxidants and biofilm itself acts as an oxidant trap. The capacity of both EPSs and biofilm to support P. aeruginosa infection in the CF lung has been reviewed [38].
5. P. aeruginosa Induces and Assimilates Host Itaconate to Cause Long-Term Disease
The host has numerous pathways to mitigate the generation of oxidants, centering around the transcription factor Nrf2 and its many downstream targets that promote the anti-oxidant response [39]. One of the metabolites that is released into the airway in response to infection is itaconate, which activates Nrf2 signaling under LPS stress [40]. Itaconate is a dicarboxylate, structurally similar to both succinate and other TCA cycle determinants and a major metabolite found in the CF airway [41]. In addition to its inhibition of macrophage SDH, itaconate also blocks glycolysis by altering the enzymatic function of both aldolase [42] and glyceraldehyde 3-phosphate dehydrogenase [43] (Figure 2). Itaconate functions as a major immuno-regulatory molecule that resolves inflammation by modulating macrophage metabolism. Itaconate also dampens IL-1β release by blocking NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) activation [44]. These effects seem to be mediated through NLRP3 decarboxypropylation on cysteine 548 (C548), which is expected to reduce NLRP3 interaction with NEK7, a major inflammasome regulator [44].
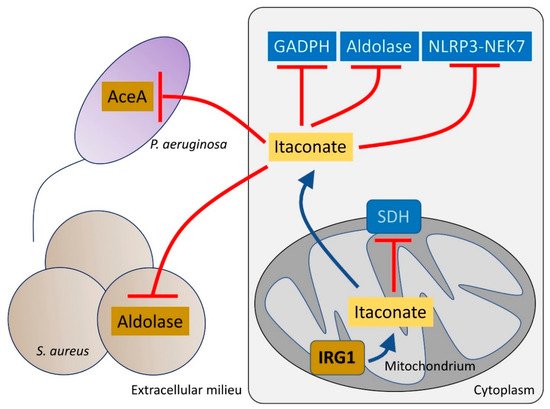
Figure 2. Itaconate controls both host and bacteria metabolism. Itaconate, synthetized by mitochondrial IRG1, inhibits host cell metabolism at different levels. Itaconate can block GADPH, aldolase and the NLRP3-NEK7 complex, which participate in pro-inflammatory signaling. Itaconate also interferes with SDH function, which is required to promote IL-1β synthesis. Once secreted, itaconate blocks the glyoxylate shunt pathway in P. aeruginosa by blocking aceA activity. In S. aureus, itaconate inhibits aldolase, suppressing glycolysis and bacterial proliferation.
Itaconate is abundantly produced by macrophages and the host airway after infection with P. aeruginosa [21][41]. Itaconate is toxic to many bacterial species, such as Staphylococcus aureus, Mycobacterium tuberculosis and Legionella pneumophila [45][46][47] targeting the activity of both isocitrate lyase (aceA) [48] and aldolase, major metabolic nodes that control the function of the anti-oxidant glyoxylate shunt [49][50] and glycolysis, respectively (Figure 2). However, several important airway pathogens, including P. aeruginosa, M. tuberculosis and Aspergillus species can also metabolize itaconate [51].
CF-adapted strains of P. aeruginosa demonstrated adaptation to itaconate using it as a carbon source, instead of succinate [41]. P. aeruginosa harbor three genes devoted to itaconate metabolism: namely, ict, ich and ccl. Expression of these genes is upregulated in response to itaconate, and this loci catabolizes itaconate to produce acetyl-CoA and pyruvate, which fuel OXPHOS function, energy production and generation of biofilm [41][51]. Itaconate is activated for degradation by ict, which produces itaconyl-CoA. Then, in a two-steps reaction ich first transforms itaconyl-CoA into its isomer mesaconyl-CoA to then hydrate it to form (S)-citramalyl-CoA. Finally, ccl breaks down (S)-citramalyl-CoA to acetyl-CoA and pyruvate, proving the bacteria with pro-energetic intermediates. Clinical isolates from chronic infection, in which itaconate is plentiful, become adapted to both induce and prefer itaconate metabolism, as the clinical strains become impaired in their ability to infect Irg1-/- mice [41]. Interestingly, pneumonia caused by a laboratory PAO1 strain, which prefers succinate over itaconate, is independent of host Irg1 function, illustrating how in vivo adaptation modulates both the immunostimulatory and metabolic preferences of these organisms.
References
- Mayer-Hamblett, N.; Ramsey, B.W.; Kulasekara, H.D.; Wolter, D.J.; Houston, L.S.; Pope, C.E.; Kulasekara, B.R.; Armbruster, C.; Burns, J.L.; Retsch-Bogart, G.; et al. Pseudomonas aeruginosa Phenotypes Associated with Eradication Failure in Children With Cystic Fibrosis. Clin. Infect. Dis. 2014, 59, 624–631.
- Folkesson, A.; Jelsbak, L.; Yang, L.; Johansen, H.K.; Ciofu, O.; Høiby, N.; Molin, S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: An evolutionary perspective. Nat. Rev. Genet. 2012, 10, 841–851.
- Magalhães, A.P.; Lopes, S.P.; Pereira, M.O. Insights into Cystic Fibrosis Polymicrobial Consortia: The Role of Species Interactions in Biofilm Development, Phenotype, and Response to In-Use Antibiotics. Front. Microbiol. 2017, 7, 2146.
- Filkins, L.M.; O’Toole, G.A. Cystic Fibrosis Lung Infections: Polymicrobial, Complex, and Hard to Treat. PLoS Pathog. 2015, 11, e1005258.
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531.
- Riquelme, S.A.; Lung, T.W.F.; Prince, A. Pulmonary Pathogens Adapt to Immune Signaling Metabolites in the Airway. Front. Immunol. 2020, 11, 385.
- Aujoulat, F.; Roger, F.; Bourdier, A.; Lotthé, A.; Lamy, B.; Marchandin, H.; Jumas-Bilak, E. From Environment to Man: Genome Evolution and Adaptation of Human Opportunistic Bacterial Pathogens. Genes 2012, 3, 191–232.
- Shrout, J.D.; Chopp, D.L.; Just, C.L.; Hentzer, M.; Givskov, M.; Parsek, M.R. The impact of quorum sensing and swarming motility on Pseudomonas aeruginosa biofilm formation is nutritionally conditional. Mol. Microbiol. 2006, 62, 1264–1277.
- Winstanley, C.; O’Brien, S.; Brockhurst, M. Pseudomonas aeruginosa Evolutionary Adaptation and Diversification in Cystic Fibrosis Chronic Lung Infections. Trends Microbiol. 2016, 24, 327–337.
- Wong, A.; Rodrigue, N.; Kassen, R. Genomics of Adaptation during Experimental Evolution of the Opportunistic Pathogen Pseudomonas aeruginosa. PLoS Genet. 2012, 8, e1002928.
- Bochner, B.R. Global phenotypic characterization of bacteria. FEMS Microbiol. Rev. 2008, 33, 191–205.
- Collier, D.N.; Hager, P.W.; Phibbs, P.V. Catabolite repression control in the Pseudomonads. Res. Microbiol. 1996, 147, 551–561.
- Görke, B.; Stülke, J. Carbon catabolite repression in bacteria: Many ways to make the most out of nutrients. Nat. Rev. Genet. 2008, 6, 613–624.
- Rojo, F. Carbon catabolite repression inPseudomonas: Optimizing metabolic versatility and interactions with the environment. FEMS Microbiol. Rev. 2010, 34, 658–684.
- Sonnleitner, E.; Abdou, L.; Haas, D. Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2009, 106, 21866–21871.
- Wolff, J.A.; MacGregor, C.H.; Eisenberg, R.C.; Phibbs, P.V.J. Isolation and characterization of catabolite repression control mutants of Pseudomonas aeruginosa PAO. J. Bacteriol. 1991, 173, 4700–4706.
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662.
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242.
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13.
- Murphy, M.P.; O’Neill, L.A. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784.
- Riquelme, S.A.; Lozano, C.; Moustafa, A.M.; Liimatta, K.; Tomlinson, K.L.; Britto, C.; Khanal, S.; Gill, S.K.; Narechania, A.; Azcona-Gutiérrez, J.M.; et al. CFTR-PTEN–dependent mitochondrial metabolic dysfunction promotes Pseudomonas aeruginosa airway infection. Sci. Transl. Med. 2019, 11, eaav4634.
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A Secreted PTEN Phosphatase That Enters Cells to Alter Signaling and Survival. Science 2013, 341, 399–402.
- Liang, H.; He, S.; Yang, J.; Jia, X.; Wang, P.; Chen, X.; Zhang, Z.; Zou, X.; McNutt, M.A.; Shen, W.H.; et al. PTENα, a PTEN Isoform Translated through Alternative Initiation, Regulates Mitochondrial Function and Energy Metabolism. Cell Metab. 2014, 19, 836–848.
- Ortega-Molina, A.; Serrano, M. PTEN in cancer, metabolism, and aging. Trends Endocrinol. Metab. 2013, 24, 184–189.
- Riquelme, S.A.; Hopkins, B.D.; Wolfe, A.L.; DiMango, E.; Kitur, K.; Parsons, R.; Prince, A. Cystic Fibrosis Transmembrane Conductance Regulator Attaches Tumor Suppressor PTEN to the Membrane and Promotes Anti Pseudomonas aeruginosa Immunity. Immunity 2017, 47, 1169–1181.e7.
- Yehia, L.; Keel, E.; Eng, C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020, 71, 103–116.
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.-C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166.
- Riquelme, S.A.; Ahn, D.; Prince, A. Pseudomonas aeruginosa and Klebsiella pneumoniae Adaptation to Innate Immune Clearance Mechanisms in the Lung. J. Innate Immun. 2018, 10, 442–454.
- Keiser, N.W.; Birket, S.E.; Evans, I.A.; Tyler, S.R.; Crooke, A.K.; Sun, X.; Zhou, W.; Nellis, J.R.; Stroebele, E.K.; Chu, K.K.; et al. Defective Innate Immunity and Hyperinflammation in Newborn Cystic Fibrosis Transmembrane Conductance Regulator–Knockout Ferret Lungs. Am. J. Respir. Cell Mol. Biol. 2015, 52, 683–694.
- Sun, X.; Sui, H.; Fisher, J.T.; Yan, Z.; Liu, X.; Cho, H.-J.; Joo, N.S.; Zhang, Y.; Zhou, W.; Yi, Y.; et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Investig. 2010, 120, 3149–3160.
- Sun, X.; Yi, Y.; Yan, Z.; Rosen, B.H.; Liang, B.; Winter, M.C.; Evans, T.I.A.; Rotti, P.G.; Yang, Y.; Gray, J.; et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci. Transl. Med. 2019, 11, eaau7531.
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480.
- Scholte, B.J.; Horati, H.; Veltman, M.; Vreeken, R.J.; Garratt, L.W.; Tiddens, H.A.; Janssens, H.M.; Stick, S. Oxidative stress and abnormal bioactive lipids in early cystic fibrosis lung disease. J. Cyst. Fibros. 2019, 18, 781–789.
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.L.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Mantovani, M.P.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875.
- Groitl, B.; Dahl, J.-U.; Schroeder, J.W.; Jakob, U. Pseudomonas aeruginosa defense systems against microbicidal oxidants. Mol. Microbiol. 2017, 106, 335–350.
- Shi, X.; Gao, Z.; Lin, Q.; Zhao, L.; Ma, Q.; Kang, Y.; Yu, J. Meta-analysis Reveals Potential Influence of Oxidative Stress on the Airway Microbiomes of Cystic Fibrosis Patients. Genom. Proteom. Bioinform. 2019, 17, 590–602.
- Ciofu, O.; Tolker-Nielsen, T. Tolerance and Resistance of Pseudomonas aeruginosa Biofilms to Antimicrobial Agents—How P. aeruginosa Can Escape Antibiotics. Front. Microbiol. 2019, 10, 913.
- Franklin, M.J.; Nivens, D.E.; Weadge, J.T.; Howell, P.L. Biosynthesis of the Pseudomonas aeruginosa Extracellular Polysaccharides, Alginate, Pel, and Psl. Front. Microbiol. 2011, 2, 167.
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426.
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117.
- Riquelme, S.A.; Liimatta, K.; Lung, T.W.F.; Fields, B.; Ahn, D.; Chen, D.; Lozano, C.; Sáenz, Y.; Uhlemann, A.-C.; Kahl, B.C.; et al. Pseudomonas aeruginosa Utilizes Host-Derived Itaconate to Redirect Its Metabolism to Promote Biofilm Formation. Cell Metab. 2020, 31, 1091–1106.e6.
- Qin, W.; Qin, K.; Zhang, Y.; Jia, W.; Chen, Y.; Cheng, B.; Peng, L.; Chen, N.; Liu, Y.; Zhou, W.; et al. S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat. Chem. Biol. 2019, 15, 983–991.
- Liao, S.-T.; Han, C.; Xu, D.-Q.; Fu, X.-W.; Wang, J.-S.; Kong, L.-Y. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat. Commun. 2019, 10, 1–11.
- Hooftman, A.; Angiari, S.; Hester, S.; Corcoran, S.E.; Runtsch, M.C.; Ling, C.; Ruzek, M.C.; Slivka, P.F.; McGettrick, A.F.; Banahan, K.; et al. The Immunomodulatory Metabolite Itaconate Modifies NLRP3 and Inhibits Inflammasome Activation. Cell Metab. 2020, 32, 468–478.e7.
- Naujoks, J.; Tabeling, C.; Dill, B.; Hoffmann, C.; Brown, A.; Kunze, M.; Kempa, S.; Peter, A.; Mollenkopf, H.-J.; Dorhoi, A.; et al. IFNs Modify the Proteome of Legionella-Containing Vacuoles and Restrict Infection Via IRG1-Derived Itaconic Acid. PLoS Pathog. 2016, 12, e1005408.
- Tomlinson, K.L.; Lung, T.W.F.; Dach, F.; Annavajhala, M.K.; Gabryszewski, S.J.; Groves, R.A.; Drikic, M.; Francoeur, N.J.; Sridhar, S.H.; Smith, M.L.; et al. Staphylococcus aureus induces an itaconate-dominated immunometabolic response that drives biofilm formation. Nat. Commun. 2021, 12, 1–13.
- Wang, H.; Fedorov, A.A.; Fedorov, E.V.; Hunt, D.M.; Rodgers, A.; Douglas, H.L.; Garza-Garcia, A.; Bonanno, J.B.; Almo, S.C.; de Carvalho, L.P.S. An essential bifunctional enzyme in Mycobacterium tuberculosis for itaconate dissimilation and leucine catabolism. Proc. Natl. Acad. Sci. USA 2019, 116, 15907–15913.
- Rittenhouse, J.W.; McFadden, B.A. Inhibition of isocitrate lyase from Pseudomonas indigofera by itaconate. Arch. Biochem. Biophys. 1974, 163, 79–86.
- Ahn, S.; Jung, J.; Jang, I.-A.; Madsen, E.L.; Park, W. Role of Glyoxylate Shunt in Oxidative Stress Response. J. Biol. Chem. 2016, 291, 11928–11938.
- Ha, S.; Shin, B.; Park, W. Lack of glyoxylate shunt dysregulates iron homeostasis in Pseudomonas aeruginosa. Microbiology 2018, 164, 587–599.
- Sasikaran, J.; Ziemski, M.; Zadora, P.K.; Fleig, A.; Berg, I. Bacterial itaconate degradation promotes pathogenicity. Nat. Chem. Biol. 2014, 10, 371–377.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
964
Revisions:
2 times
(View History)
Update Date:
06 Aug 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No