 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Youichirou Higashi | + 1364 word(s) | 1364 | 2021-07-26 08:19:43 | | | |
| 2 | Vivi Li | Meta information modification | 1364 | 2021-08-06 04:00:31 | | |
Video Upload Options
Stroke is a major cause of death worldwide, leading to serious disability. Post-ischemic injury, especially in the cerebral ischemia-prone hippocampus, is a serious problem, as it contributes to vascular dementia. Many studies have shown that in the hippocampus, ischemia/reperfusion induces neuronal death through oxidative stress and neuronal zinc (Zn2+) dyshomeostasis. Glutathione (GSH) plays an important role in protecting neurons against oxidative stress as a major intracellular antioxidant. In addition, the thiol group of GSH can function as a principal Zn2+ chelator for the maintenance of Zn2+ homeostasis in neurons. These lines of evidence suggest that neuronal GSH levels could be a key factor in post-stroke neuronal survival. In neurons, excitatory amino acid carrier 1 (EAAC1) is involved in the influx of cysteine, and intracellular cysteine is the rate-limiting substrate for the synthesis of GSH. Recently, several studies have indicated that cysteine uptake through EAAC1 suppresses ischemia-induced neuronal death via the promotion of hippocampal GSH synthesis in ischemic animal models.
1. Introduction
2. Protective Roles of Glutathione (GSH) in Ischemia-Induced Hippocampal Injury
2.1. Anti-Oxidative Role of GSH in Ischemia-Induced Oxidative Stress
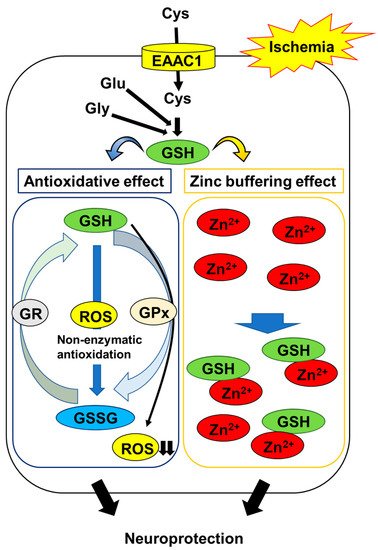
2.2. GSH Protects Neurons from Ischemia-Induced Disruption of Intracellular Zn2+ Homeostasis
References
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480.
- Feigin, V.L.; Krishnamurthi, R. Public health strategies could reduce the global stroke epidemic. Lancet Neurol. 2010, 9, 847–848.
- Izquierdo, I.; Medina, J.H. Memory formation: The sequence of biochemical events in the hippocampus and its connection to activity in other brain structures. Neurobiol. Learn. Mem. 1997, 68, 285–316.
- Schaapsmeerders, P.; van Uden, I.W.M.; Tuladhar, A.M.; Maaijwee, N.A.M.; van Dijk, E.J.; Rutten-Jacobs, L.C.A.; Arntz, R.M.; Schoonderwaldt, H.C.; Dorresteijn, L.D.A.; de Leeuw, F.E.; et al. Ipsilateral hippocampal atrophy is associated with long-term memory dysfunction after ischemic stroke in young adults. Hum. Brain Mapp. 2015, 36, 2432–2442.
- Oliver, C.N.; Starke-Reed, P.E.; Stadtman, E.R.; Liu, G.J.; Carney, J.M.; Floyd, R.A. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5144–5147.
- Rodrigo, R.; Fernández-Gajardo, R.; Gutiérrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeutic opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714.
- Takeda, A. Zinc homeostasis and functions of zinc in the brain. Biometals 2001, 14, 343–351.
- Koh, J.Y.; Suh, S.W.; Gwag, B.J.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996, 272, 1013–1016.
- Won, S.J.; Yoo, B.H.; Brennan, A.M.; Shin, B.S.; Kauppinen, T.M.; Berman, A.E.; Swanson, R.A.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and exacerbates neuronal injury after transient cerebral ischemia. J. Neurosci. 2010, 30, 15409–15418.
- Aratake, T.; Higashi, Y.; Hamada, T.; Ueba, Y.; Shimizu, T.; Shimizu, S.; Yawata, T.; Ueba, T.; Saito, M. The role of diurnal fluctuations in excitatory amino acid carrier 1 levels in post-ischemic hippocampal Zn2+ accumulation. Exp. Neurol. 2021, 336, 113538.
- Kapoor, M.; Sharma, S.; Sandhir, R.; Nehru, B. Temporal changes in physiological and molecular markers in various brain regions following transient global ischemia in rats. Mol. Biol. Rep. 2019, 46, 6215–6230.
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Park, K.H.; Song, H.K.; Choi, H.C.; Suh, S.W. Effects of protocatechuic acid (PCA) on global cerebral ischemia-induced hippocampal neuronal death. Int. J. Mol. Sci. 2018, 19, 1420.
- Love, S. Oxidative stress in brain ischemia. Brain Pathol. 1999, 9, 119–131.
- Nikonenko, A.G.; Radenovic, L.; Andjus, P.R.; Skibo, G.G. Structural features of ischemic damage in the hippocampus. Anat. Rec. 2009, 292, 1914–1921.
- Wang, Q.; Tompkins, K.D.; Simonyi, A.; Korthuis, R.J.; Sun, A.Y.; Sun, G.Y. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006, 1090, 182–189.
- Galano, A.; Alvarez-Idaboy, J.R. Glutathione: Mechanism and kinetics of its non-enzymatic defense action against free radicals. RSC Adv. 2011, 1, 1763–1771.
- Orian, L.; Cozza, G.; Maiorino, M.; Toppo, S.; Ursini, F. The catalytic mechanism of glutathione peroxidases (Chapter 3). In Glutathione, 1st ed.; Flohé, L., Ed.; CRC Press: Boca Raton, FL, USA, 2018; pp. 35–52.
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42.
- Hirrlinger, J.; Dringen, R. Multidrug resistance protein 1-mediated export of glutathione and glutathione disulfide from brain astrocytes. Methods Enzymol. 2005, 400, 395–409.
- Homolya, L.; Váradi, A.; Sarkadi, B. Multidrug resistance-associated proteins: Export pumps for conjugates with glutathione, glucuronate or sulfate. BioFactors 2003, 17, 103–114.
- Wu, T.; Yin, F.; Kong, H.; Peng, J. Germacrone attenuates cerebral ischemia/reperfusion injury in rats via antioxidative and antiapoptotic mechanisms. J. Cell. Biochem. 2019, 120, 18901–18909.
- Li, Z.; Yulei, J.; Yaqing, J.; Jinmin, Z.; Xinyong, L.; Jing, G.; Min, L. Protective effects of tetramethylpyrazine analogue Z-11 on cerebral ischemia reperfusion injury. Eur. J. Pharmacol. 2019, 844, 156–164.
- Abdel-Fattah, M.M.; Messiha, B.A.S.; Mansour, A.M. Modulation of brain ACE and ACE2 may be a promising protective strategy against cerebral ischemia/reperfusion injury: An experimental trial in rats. Naunyn-Schmiedebergs Arch. Pharmacol. 2018, 391, 1003–1020.
- Yabuki, Y.; Fukunaga, K. Oral administration of glutathione improves memory deficits following transient brain ischemia by reducing brain oxidative stress. Neuroscience 2013, 250, 394–407.
- Anderson, M.F.; Nilsson, M.; Eriksson, P.S.; Sims, N.R. Glutathione monoethyl ester provides neuroprotection in a rat model of stroke. Neurosci. Lett. 2004, 354, 163–165.
- Won, S.J.; Kim, J.E.; Cittolin-Santos, G.F.; Swanson, R.A. Assessment at the single-cell level identifies neuronal glutathione depletion as both a cause and effect of ischemia-reperfusion oxidative stress. J. Neurosci. 2015, 35, 7143–7152.
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569.
- Frederickson, C.J. Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 1989, 31, 145–238.
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1716–1721.
- Danscher, G.; Howell, G.; Pérez-Clausell, J.; Hertel, N. The dithizone, Timm’s sulphide silver and the selenium methods demonstrate a chelatable pool of zinc in CNS. A proton activation (PIXE) analysis of carbon tetrachloride extracts from rat brains and spinal cords intravitally treated with dithizone. Histochemistry 1985, 83, 419–422.
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791.
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462.
- Frederickson, C.J.; Giblin, L.J.; Krezel, A.; McAdoo, D.J.; Mueller, R.N.; Zeng, Y.; Balaji, R.V.; Masalha, R.; Thompson, R.B.; Fierke, C.A.; et al. Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp. Neurol. 2006, 198, 285–293.
- Lee, J.Y.; Kim, J.H.; Palmiter, R.D.; Koh, J.Y. Zinc released from metallothionein-III may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp. Neurol. 2003, 184, 337–347.
- Koh, J.Y.; Lee, S.J. Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol. Brain 2020, 13, 116.
- Krężel, A.; Maret, W. The functions of metamorphic metallothioneins in zinc and copper metabolism. Int. J. Mol. Sci. 2017, 18, 1237.
- Medvedeva, Y.V.; Ji, S.G.; Yin, H.Z.; Weiss, J.H. Differential vulnerability of CA1 versus CA3 pyramidal neurons after ischemia: Possible relationship to sources of Zn2+ accumulation and its entry into and prolonged effects on mitochondria. J. Neurosci. 2017, 37, 726–737.
- Maret, W. Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc. Natl. Acad. Sci. USA 1994, 91, 237–241.
- Wang, T.; Zheng, W.; Xu, H.; Zhou, J.M.; Wang, Z.Y. Clioquinol inhibits zinc-triggered caspase activation in the hippocampal CA1 region of a global ischemic gerbil model. PLoS ONE 2010, 5, e11888.
- Chen, C.J.; Liao, S.L. Zinc toxicity on neonatal cortical neurons: Involvement of glutathione chelation. J. Neurochem. 2003, 85, 443–453.
- Hong, D.K.; Kho, A.R.; Lee, S.H.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, M.K.; Park, K.H.; Lim, M.S.; Choi, B.Y.; et al. Transient receptor potential melastatin 2 (TRPM2) inhibition by antioxidant, n-acetyl-l-cysteine, reduces global cerebral ischemia-induced neuronal death. Int. J. Mol. Sci. 2020, 21, 6026.
- Mize, C.E.; Langdon, R.G. Hepatic glutathione reductase. I. Purification and general kinetic properties. J. Biol. Chem. 1962, 237, 1589–1595.
- Splittgerber, A.G.; Tappel, A.L. Inhibition of glutathione peroxidase by cadmium and other metal ions. Arch. Biochem. Biophys. 1979, 197, 534–542.



