 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Karen C. Glass | + 4090 word(s) | 4090 | 2021-07-29 12:35:56 | | | |
| 2 | Beatrix Zheng | + 760 word(s) | 4850 | 2021-08-07 16:35:57 | | |
Video Upload Options
This review provides an in depth analysis of the role of bromodomain-containing proteins in cancer development. As readers of acetylated lysine on nucleosomal histones, bromodomain proteins are poised to activate gene expression, and often promote cancer progression. We examined changes in gene expression patterns that are observed in bromodomain-containing proteins and associated with specific cancer types. We also mapped the protein–protein interaction network for the human bromodomain-containing proteins, discuss the cellular roles of these epigenetic regulators as part of nine different functional groups, and identify bromodomain-specific mechanisms in cancer development. Lastly, we summarize emerging strategies to target bromodomain proteins in cancer therapy, including those that may be essential for overcoming resistance. Overall, this review provides a timely discussion of the different mechanisms of bromodomain-containing proteins in cancer, and an updated assessment of their utility as a therapeutic target for a variety of cancer subtypes.
1. Introduction
Cancer heterogeneity presents major challenges for the development of personalized treatments [1][2][3][4]. Current precision therapies used in the treatment of cancer are designed to exploit a variety of different biological entities characteristic to individual cancer types, such as activated protein kinases, estrogen receptor, and defective DNA repair enzymes [4][5][6]. Understanding the mechanistic details of cancer biology is critical for improving diagnostic tools and for developing new therapeutic interventions. A comprehensive understanding of cancer requires interpretation of molecular intricacies at multiple levels such as genomic, epigenomic, transcriptomic, proteomic, and metabolomic data. With the advent of high-throughput technologies, the availability of multi-omics data has revolutionized our understanding of the disease process and has created new avenues for integrated system-level approaches.
Epigenetic mechanisms are increasingly being recognized as central to the development and progression of cancer. Epigenetic changes are defined as heritable non-genetic mechanisms that impact gene expression [7]. These mechanisms include DNA methylation, post-translational histone modifications (PTMs), and non-coding RNAs (i.e., microRNAs and long non-coding RNA s), which play fundamental roles in essentially all nuclear processes involving DNA, including transcription, DNA replication, and DNA repair [8][9][10]. Improved understanding of the epigenetic mechanisms underlying cancer etiology has resulted in the identification of a number of molecular targets and the development of novel therapeutics and prognostic biomarkers. Many epigenetic inhibitors have emerged as attractive anti-cancer agents in pre-clinical studies [11]. In particular, the recent advent of small-molecule inhibitors that target bromodomains has provided critical insight into our understanding of the biological mechanisms of bromodomain proteins in cancer. Research in this area has focused on the development of inhibitors for the bromodomain and extra-terminal motif (BET) bromodomains; however, more recently, inhibitors targeting the non-BET bromodomains have emerged. These inhibitors have provided new insights into the cellular function of non-BET bromodomain proteins, and our increasing knowledge of bromodomain structure and function has shed light on the structural aspects of the selective histone recognition activities of all bromodomain proteins. The rationale design of a second generation of compounds has produced bromodomain inhibitors that selectively target individual bromodomain proteins, including non-BET bromodomain proteins [12]. However, a great deal remains to be understood regarding the role bromodomain proteins play in cancer progression. Understanding their expression and interaction profiles, and their regulatory roles in chromatin modifiers, could provide additional insight into bromodomain-dependent mechanisms in cancer [13]. The role of bromodomain inhibitors across a variety of cancers may yet be important to refine personalized medicine in cancer treatment.
In this review, we systematically evaluate bromodomain-containing proteins as individual entities of a larger family of epigenetic regulators, highlighting recent advances in our understanding of how recognition of acetylated lysine by the bromodomain influences protein function. Our goal is to outline the function of bromodomain proteins in different biological contexts and to provide insights on the functional role of the bromodomain in these processes from the lessons learned by examining the cellular effects of small-molecule bromodomain inhibitors as potential anti-cancer agents.
2. Bromodomains Are Histone Lysine Acetylation Reader Domains
Bromodomains are protein interaction modules that “read” ε-N-lysine acetylation (Kac) marks [14]. There are other known protein domains that recognize and bind to Kac, including the plant homeodomain (PHD finger) and the Yaf9, ENL, AF9, Taf14, and Sas5 (YEATS) domains [15][16]. However, their primary targets encompass additional PTMs including methylated and crotonylated lysine, respectively [17][18]. Through their ability to read a variety of different acetyllysine modifications present on each of the different core and variant histones, bromodomains play a critical role in orchestrating protein and DNA complexes at chromatin. Owing to their central role in chromatin function, bromodomain-containing proteins have been attributed to play prominent roles in the development and progression of a spectrum of diseases, including cancer and other cardiovascular, metabolic, inflammatory, neurologic, and musculoskeletal diseases [19].
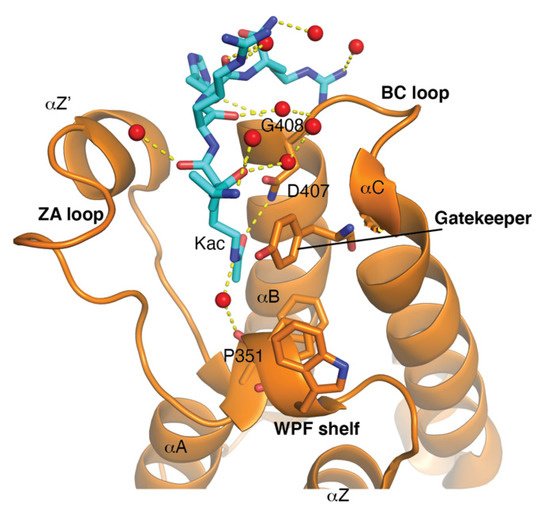
The bromodomain is a 110-amino-acid structural motif that forms of a bundle of four α-helices (αZ, αA, αB, and αC) where the interhelical αZ-αA (ZA) and αB- αC (BC) loops create a hydrophobic pocket that recognizes acetyllysine modifications. Although the primary sequence varies between bromodomains, certain residues in the BC loop region that are involved in Kac coordination are highly conserved [20]. The bromodomain structure from the general control non-depressible 5 (Gcn5p) HAT in complex with an acetylated histone H4 peptide was solved using X-ray crystallography [21]. This structure revealed that the site of Kac recognition was in the hydrophobic pocket formed between the ZA and BC loops. Inside this binding pocket, the carbonyl oxygen on the acetyl group of the lysine forms a hydrogen bond with a nitrogen in the amide group of asparagine 407 in Gcn5p ( Figure 1 ). This asparagine residue is nearly universally conserved in all bromodomains and is important for Kac recognition. Additionally, the histone H4 peptide was found to interact with several ordered water molecules that help stabilize the ligand inside the binding pocket [21].
There are a number of different Lys residues present on the N- terminus of each of the core histone proteins that are known to be acetylated: 10 in histone H2A, 16 in H2B, 13 in H3, and 9 in H4 [22]. The histone ligand binding activity towards specific Kac modifications has been examined in many of the human bromodomains found in different subfamilies [23]. Further characterization of their preferred histone ligands has been carried out using a variety of different biophysical methods that employ recombinant bromodomains and modified histone peptides, or nucleosome substrates in binding reactions [19]. Classically, methods used for ligand identification included Western blots, isothermal titration calorimetry, fluorescent polarization spectroscopy, surface plasmon resonance (SPR), and NMR. Newer, more high-throughput methods include peptide arrays [23][24] and the development of AlphaScreen-based peptide assays to detect bromodomain ligands from many different combinations of histone modifications [25][26]. For example, the ligand binding of the ATPase family AAA domain-containing 2 (ATAD2 and ATAD2B) bromodomains was recently compared using the dCypher assay developed by EpiCypher [26]. The recombinantly expressed GST-tagged bromodomains of the ATAD2 and ATAD2B paralogs were screened against 288 unique histone ligands containing single- and multiple-modified histone peptides representing PTMs found in all four of the core histones. Interestingly, although the ATAD2/B bromodomains are highly conserved and recognize similar histone ligands, it was discovered that the ATAD2B bromodomain has a much broader range of PTM histone binding partners. The ATAD2B bromodomain interacted with 39 ligands from histones H4 and H2A, compared to ATAD2A, which bound to 11 ligands on histone H4. Both of these bromodomains showed a strong preference for histone H4, recognizing acetylated lysine at residues 5, 8, and 12, similar to what was previously reported for ATAD2 using time-resolved fluorescence resonance energy transfer (TR-FRET) methodology [27]. Using isothermal titration calorimetry (ITC) and modified peptides, Lloyd et al. determined the dissociation constants (K D ) for mono- and di-acetylated histone ligands, demonstrating that the ATAD2B bromodomain preferentially recognizes histone H5K5ac (5.2 ± 1.0 μM), followed by several di-acetylated histone peptides including H4K5acK12ac (18.7 ± 0.9 μM). Other bromodomains have also been shown to recognize di-acetylation modifications, including bromodomain and PHD finger containing 1 (BRPF1), bromodomain containing 9 (BRD9), TATA-box binding protein associated factor 1 (TAF1), and the BD1/BD2 bromodomains in the BET subfamily, suggesting a cooperative role of local sites for ligand binding [23][28][29][30].
Over the last few years, proteomics studies have identified additional acyl modifications found on histone lysine residues that appear at a much lower frequency compared to acetylation [31][32][33]. These include histone lysine propionylation, butyrylation, crotonylation, succinylation, malonylation, 5-hyroxylation, and N-formylation (Kpr, Kbr, Kcr, Ksu, Kmal, Khy, and Kfo, respectively) [31][32][33][34][35]. The bromodomains of BRD4 were shown to recognize Kpr and Kbu, with significantly reduced binding affinities compared to Kac at the same residue [36]. Similarly, 49 human bromodomains were screened for binding to peptides bearing related acyl PTMs, including Kac, Kpr, Kbu, Kcr, Kfo, and Ksu [29]. While Kpr commonly bound to the bromodomains screened, only three bromodomains could bind Kbu (CECR2 histone acetyl-lysine reader, BRD9, and TAF1/L), and a single bromodomain showed binding to Kcr at reduced affinity (TAF1/L) [29]. None of the bromodomains tested showed affinity for Ksu using the peptide array platform. There are also reports of bromodomain interactions with acetylated non-histone proteins, which highlights the complex role that bromodomain proteins play in biology [37][38][39][40]. Lastly, despite the large number of PTMs found on histones, our understanding of how these PTM combinations effect chromatin recognition is very limited. However, this knowledge will be essential for deciphering the comprehensive nature of bromodomain ligand recognition in the context of the epigenetic landscape.
3. An Interconnected Network of Functional Groups in Bromodomain Proteins
While the human bromodomains have been divided into eight subfamilies based on their structural features, they are found in a wide range of proteins with diverse catalytic and scaffolding functions. Bromodomains often act in concert with other functional modules present in the same proteins, and in their associated proteins complexes. As bromodomain-containing proteins assemble into larger complexes to confer context-specific activity, the systematic analysis of protein–protein interactions (PPIs) is useful to summarize potential functional pathways associated with the cellular roles of these chromatin reader proteins. The availability of high-throughput proteomic techniques, such as Affinity Purification coupled to Mass Spectrometry (AP-MS) [41], BioID [42], and recently developed TurboID [43] enabled us to collect protein–protein interaction data from public databases including the Biological General Repository for Interaction Datasets (BioGRID) [44] and the Human Integrated Protein–Protein Interaction reference (HIPPIE) [45], as well as the newly reported interactions for BETs [37]. The global interaction network of all human bromodomain proteins collected from the above resources is shown in Figure 2 A, and listed in Table S1 . This interaction network shows a highly interconnected map of PPIs of bromodomain proteins grouped according to their function-based classifications. Different functional classes of bromodomain proteins cluster together in the network, based on shared interactions, and the formation of related functional complexes. For example, the BET family of bromodomain proteins, including BRD2, BRD3, BRD4, and BRDT (Bromodomain Testis associated) shown in blue, cluster together as they are well known to share many common interaction partners [37][46]. Similarly, the HAT enzymes including the p300 and cyclic AMP response element-binding protein (p300/CBP), and the monocytic leukemia zinc-finger protein and MOZ-related factor (MOZ/MORF) groups cluster together in this network. Additionally, we also identified a subnetwork of bromodomain–bromodomain protein interactions that demonstrates how the bromodomain-containing proteins interact with each other (Figure 2 B). Thus, functional clustering based on their PPI networks provides a succinct way to represent the many inter-connected roles for bromodomain-containing proteins. Below, we discuss nine different functional groups of bromodomain proteins, highlighting recent insights into bromodomain-specific mechanisms in cancer development.
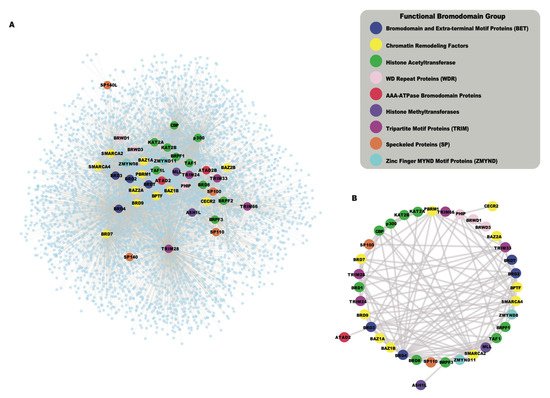
More recently, a systematic proteomic approach was used to analyze the overall protein interactions of BET proteins. Lambert et al. carried out affinity purification on the BET bromodomains followed by mass spectrometry (AP-MS) before and after the addition of the pan-BET inhibitor JQ1 [37]. Quantitative analysis of 603 unique interacting proteins defined three distinct sets of BET protein interactions including those that occur through the canonical acetyllysine binding pocket to recognize acetylated histone and non-histone proteins, as well as acetylation-independent interactions via the extra-terminal domain. This study examined the human proteome to identify di-acetyllysine motifs on histone and non-histone proteins, and further characterized the interaction of BET bromodomains with these Kac-XX-Kac motifs using a combination of biophysical, structural, and cell biology approaches. Importantly, they identified several new non-histone interactions, some that were increased upon addition of JQ1, and found the ET domain provides an important protein recruitment platform. They also demonstrated that BRD3 is a negative regulator of cellular proliferation via regulation of ribosomal RNA production. This comprehensive study is the first to clearly demonstrate how small-molecule inhibitors targeting specific bromodomain containing proteins can be effectively used as tool compounds to elucidate the biological functions of bromodomains as well as inhibitor action.
The speckled protein (SP) family of bromodomain proteins includes SP100, SP110, SP140, and the SP140-like protein (SP140L) [48]. The SP proteins have an N-terminal caspase activation and recruitment domain (CARD), which is typically involved in oligomerization, and is found in a wide variety of proteins that often contribute to apoptosis, the inflammatory response, or immunogenic signaling [49]. In SP proteins, the CARD domain is speculated to be involved in homodimerization, but its role is poorly characterized in terms of its function in immune cells [48]. The SP family proteins also contain several functional domains that implicate them as chromatin readers. The SAND domain (named according to proteins that have it: SP100, Aire, NucP41/P75, and DEAF) has been shown to bind to DNA and mediate protein–protein interactions [50][51]. The C-terminal PHD finger and bromodomain function in histone recognition.
Similar to SP100, the SP140 and SP140L proteins are autoantigens in primary biliary cirrhosis, and SP140 is active in chronic lymphocytic leukemia [48]. The PHD-bromodomain cassette found in SP140 preferentially recognizes unmodified histone H3K4me0; however, its binding was disrupted in the presence of methylation at either H3K4 or H3K9 [52]. Interestingly, the SP140 protein appears to be regulated via SUMOylation, and the PHD-Bromodomain is a SUMOylation target of SUMO-1, with the PHD finger facilitating binding of the Ubc9 E2 ligase and SUMO-1 to stimulate SUMOylation of the adjacent bromodomain [53]. The SP family of proteins is involved in several aspects of the innate cellular immunity pathways [48]. Although inhibitors are not currently available for this class of bromodomain-containing proteins, there may be an opportunity to target them for immunosuppressive applications in the future.
4. Emerging Strategies to Target Bromodomain Proteins
The discovery of JQ1 and I-BET as potent and selective inhibitors for the BET family of bromodomains shifted the paradigm for the chromatin reader field. Prior to the development of JQ1, it was thought that bromodomain inhibitors would have broad activity towards all human bromodomain-containing proteins, resulting in extensive off-target effects. Instead, JQ1 was shown to selectively target the bromodomains of BRD4, BRD3, and BRD2 with nanomolar binding affinities, while also having very little activity towards non-BET bromodomains [54]. Furthermore, the use of JQ1 in a mouse model of nuclear protein in testis (NUT) midline carcinoma, which results from a chromosomal fusion of BRD4 with NUT, resulted in improved survival and tumor regression [54]. Similarly, I-BET was shown to preferentially bind to the bromodomain of BRD4, followed by BRD3, and BRD2, and disrupted their interaction with tetra-acetylated histone H4 ligands. These early studies stimulated a broad interest in understanding the structure and functions of bromodomains, and current advances include the characterization of the structures and histone ligand binding activities for nearly all of the human bromodomains [23]. Several investigations into the mechanism of action for BET bromodomain inhibitors have provided new insights into the protein interaction networks of these proteins with both histone and non-histone proteins [37]. For example, BRD3 was shown to be a binding partner of the GATA1 transcription factor via recognition of specific acetyllysine modifications, and the addition of a BRD inhibitor disrupted this interaction [39]. Importantly, BET bromodomain inhibition was identified as a therapeutic strategy in multiple myeloma, working to inhibit the transcription of the c-Myc oncoprotein, which resulted in cellular senescence and halted proliferation of leukemia cells [55]. BET inhibition has also been used to study the role of these bromodomains in HIV infection, leukemogenesis, and spermatogenesis [56][57][58]. Numerous BET inhibitors have been evaluated, or are currently undergoing evaluation, in clinical trials. Importantly, while JQ1 is an essential tool compound for studying BET bromodomain function(s), it has been extensively modified for use in the clinic. JQ1 derivatives, such as OTX015 (Birabriseb), have demonstrated greater therapeutic efficacy. Successful targeting of the BET bromodomain family spurred a global interest in the development of additional bromodomain inhibitors targeting both BET and non-BET bromodomain proteins. While the inhibition of BET bromodomains for cancer therapy has remained a highly active area of research, there are now chemical probes available to specifically target individual bromodomain-containing proteins from all eight bromodomain subfamilies [59]. Table 1 provides a current summary of promising BETi and non-BET inhibitors that show potential in the clinical setting.
| BRD Inhibitor (Molecule Images Created with JSME [60]) |
Target | Cancer Type/Results | Clinical Trial ID/Reference | |
|---|---|---|---|---|
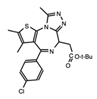 |
JQ1 | BET’s | The first generation of BET inhibitors which has proven to be a valuable tool for understanding BET BRDs in numerous cancers, but demonstrated toxicities in the clinic. As a result derivatives of JQ1 have had greater clinical success. | [27][61][62] |
 |
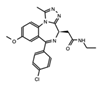
OTX015* (Birabresib) |
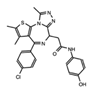
BET’s | Identifiers and resulting publications for ongoing and completed clinical trials for OTX015 as an exclusive therapy or in combination treatment. This JQ1 derivative used in combination with PROTACs has shown promise in cell models of prostate cancer, lymphoma, and leukemia. |
NCT02698176, NCT02259114, NCT01713582, NCT02698189, NCT02296476 [63][64][65][66][67][68][69] |
 |
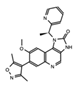
I-BET762 GSK525762A, (Molibresib) |
BET’s | Phase 1 clinical trial of this orally available compound initially showed that daily dosing with molibresib was well tolerated and showed efficacy for patients with nuclear protein in testis (NUT) carcinoma. | [28][69][70][71] |
 |
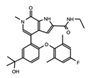
I-BET151 GSK1210151A |
BET’s | This BET inhibitor demonstrates strong anti-proliferative effects, and xenograft models indicate repression of proliferation in myeloma cells. However, this drug has not made progressed to clinical trials. | [72] |
 |
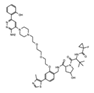
ABBV-744 | Pan-BET (Selective for the 2nd bromodo-main) |
Selective for the 2nd bromodomain of the BET-bromodomain proteins, and has demonstrated anti-proliferative effects for numerous acute myeloid leukemia and prostate cancer cell lines. | NCT04454658 [73][74] |
| Not available | ZEN-3694 | BET’s | A current phase 2 clinical trial using ZEN-3694 in combination with the enzalutamide is recruiting for castration resistant prostate cancer (CRPC), where the compound has shown efficacy in a phase 1 clinical trial. | NCT04471974 [75] |
 |
ACBI1 | SMARCA2/4 | This PROTAC degrader resulted in reduced protein levels and apoptosis of acute myeloid leukemia (AML) cells. | [76] |
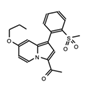 |
GSK2801 | BAZ2A/B | The selective acetyl-lysine competitive inhibitor induces apoptosis in triple negative breast cancer (TNBC) cells in combination with BET inhibitors. | [77][78] |
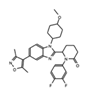 |
CCS1477 | CBP/p300 | Current Phase 1 & 2 clinical trials are recruiting patients for treatment of hematological malignancies and advanced prostate cancer. | NCT03568656, NCT04068597 |
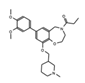 |
I-CBP112 | CBP/p300 | Combination therapy with the p300/CBP active site inhibitor (A-485) resulted in reduced p300 chromatin enrichment, and decreased expression of androgen-dependent and pro-oncogenic genes in leukemia and prostate cancer. | [79][80] |
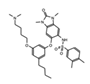 |
IACS-9571 | BRPF1/TRIM24 | This selective inhibitor has provided insights into cellular functions, and may be useful as a potential therapeutic for acute myeloid leukemia (AML) and breast cancer (BCa). | [81] |
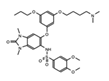 |
IACS-9571 | TRIM24 | When developed into a bifunctional degrader linked to the VHL E3 ligase, TRIM24 protein degradation resulted in a greater negative impact on proliferation in leukemia cell lines. | [82] |
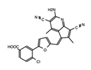 |
AM879 | ATAD2 | Treatment with AM879, prevented cell proliferation, and induced apoptosis in triple negative breast cancer (TNBC) cells. | [83] |
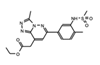 |
Bromospo-rine | Multi-BRD (BET) | Shows promise as a therapeutic for colorectal cancer (CRC) when administered in combination with 5-Fluorouracil (5-FU). | [12][84] |
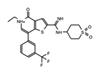 |
I-BRD9 | BRD9 | The selective inhibitor identified cancer associated and immune response genes as possible targets of BRD9 regulation in leukemia cells. | [85] |
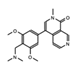 BI-7273 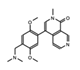 BI-9564 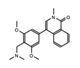 BI-7271 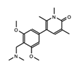 BI-7189 |
BI-7273/BI-9564 BI-7271/BI-7273/BI-7189 |
BRD9 | These small molecule inhibitors display anti-tumor activity in xenograft models of AML. | [86] |
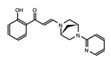 |
PFI-3 | SMARCA2/4 and PB1(5) | Treatment with PFI-3 has been shown to sensitize cancer cells to chemotherapeutic agents. | http://www.thesgc.org/chemical-probes/PFI-3, accessed on 7 July 2021 [87] |
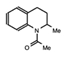 MS2126 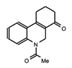 MS7972 |
MS2126/MS7972 | CBP/p300 | Cell based assays in osteosarcoma cells, demonstrated that these molecules can modulate the p53 response to DNA damage. | [88] |
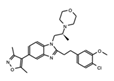 |
SGC-CBP30 | CBP/p300 | Inhibition of CBP is suggested to be a potential method for targeting transcriptional dependencies in multiple myeloma. | [89][90][91] |
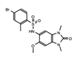 OF-1 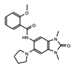 PFI-4 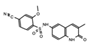 NI-57 |
OF-1, PFI-4, NI-57 | pan-BRPF | While these inhibitors have not be linked to anti-proliferative or anti-cancer therapies, they have been suggested as potential therapeutics in bone malignancies. | [92] |
Understanding the biological relevance, structural uniqueness, and clinical applications of BRD inhibition are important steps in the development of BRD inhibitors as cancer therapeutics. Although the downregulation of MYC transcription has been widely proposed as a key mechanism for BET inhibitor anti-tumor activity [55], additional mechanisms for BET inhibitor efficacy against cancer have also been reported. For example, it has been demonstrated in models of B-cell lymphoma that BET inhibition modulates the expression of pro- and anti-apoptotic BCL-2 family members to induce apoptosis through intrinsic mitochondrial apoptotic pathways [93][94]. The efficacy of BET inhibitors as anti-cancer agents has been evaluated in preclinical studies for multiple cancer types, with drugs presently at different stages of clinical trials ( Table 1 ). However, as with most targeted cancer therapies, resistance to inhibitors limits their effectiveness in patients. A variety of mechanisms underlying resistance to BET inhibitors have been reported for different tumor types. BET inhibitor resistance has been widely reported to occur through the reactivation of MYC expression. For example, in a study on the development BET inhibitor resistance in acute myeloid leukemia, Fong C et al. discovered that stimulation of the Wnt/beta-catenin pathway resulted in increased binding of beta-catenin at MYC regulatory sites where BRD4 was displaced from the chromatin. This appears to prime a subset of the leukemia stem cells for transcriptional plasticity allowing them to upregulate MYC and take over as the dominant cell type harboring BET inhibitor resistance [95]. In triple-negative breast cancer, resistance to JQ1 treatment emerged through altered epigenetic signaling of BRD4. In the resistant cells, higher levels of phosphorylated BRD4 were detected, and this led to increased binding interactions of BRD4 with MED1, resulting in decreased responsiveness to bromodomain inhibition [96]. Another study found that the voltage-dependent anion channel 1 (VDAC1) is also linked to the development of resistance to JQ1 in breast cancer [97]. More recently, a comprehensive study by Shu et al. carried out a genome-wide CRISPR screen to identify genes that contribute to the development of resistance after JQ1 treatment in triple-negative breast cancer cell lines. Importantly, they discovered additional therapeutic agents that are synergistic with JQ1 in inhibiting tumor cell growth. These included DNA-damaging agents (doxorubicin) and microtubule inhibitors (Paclitaxel/Vincristine). They found that Palbociclib had the most significant effect and worked by enhancing CDK4 inhibition-mediated G1 arrest and by destabilizing BRD2/4 via proteasomal degradation [98]. Several other studies have also found the cellular effects of bromodomain inhibition in cancer treatment to be enhanced by combination therapy. Due to their ability to intrinsic apoptosis signaling, recent reports demonstrate synergistic activity of BET inhibitors and the small-molecule BCL-2 inhibitor ABT199/venetoclax in killing MYC -driven B-cell lymphoma cells [94][99][100]. For example, the combined inhibition of both BET bromodomains and HDAC enzymes improved the efficacy of either drug class alone [101][102]. This led to the prediction that using bromodomain inhibitors in addition to the standard therapy may reduce the development of resistance in some cancers [103]. Combinations of epigenetic therapeutics have also proven successful in the treatment of acute myeloid leukemia [104], multiple myeloma [105], pancreatic cancer [106], ovarian cancer [107], and in breast cancer [108]. One mechanism for this synergy has been outlined through the dual inhibition of BET bromodomains in combination with poly[adenosine diphosphate (ADP)-ribose] polymerase inhibitors (PARPi). The addition of a bromodomain inhibitor blocks the homologous recombination (HR) DNA repair pathway in addition to the base excision repair pathway, sensitizing HR proficient cancers [109].
Another therapeutic strategy that has shown great promise to improve BRD inhibitor efficacy is the targeted degradation of bromodomain-containing proteins. Proteolysis-targeting chimeras (PROTACs) were first developed in 2001 by Sakamoto et al., in order to direct disease causing proteins for ubiquitin-dependent degradation by the proteosome [110]. The key feature of the chimeric molecule is the linker, which contains a BRD ligand mimic/small molecule on one end, and an E3 ligase recognition domain on the other. Thus, thalidomide derivatives will bring the bromodomain-containing protein of interest to the Cereblon E3 ligase, while a short peptide sequence from Hypoxia-inducible factor 1 (HIF1) will recruit the VHL E3 ligase. This has been done successfully with BRD4, BRD9, TRIM24, and PCAF/ GCN5 [111][112][113][114]. A BRD9 specific degrader was developed as a tool compound to study the function of this bromodomain-containing protein [114], and it was demonstrated that degradation of BRD9 triggered downregulation of oncogenic programs that contribute to the development of synovial sarcoma/soft tissue tumors [115]. A similar approach was used with TRIM24, where the protein is targeted for selective degradation by chemically conjugating the small-molecule inhibitor IACS-9571 to the Von Hippel–Lindau (VHL) E3 ubiquitin ligase [112]. Degradation of the entire TRIM24 protein was shown to have a more immediate and longer-lasting impact on cellular proliferation than BRD inhibition alone, particularly in leukemia cell lines [112].
The advantage of using PROTACs to degrade bromodomain-containing proteins is that these proteins are often targeted to the chromatin via multiple domains. The result is that some BRD inhibitors are not as effective as expected [116]. For example, BRD inhibitors developed for SMARCA2/4 did not produce the expected anti-proliferative effects [117][118]. Thus, degraders to disrupt the formation of active SMARCA2/4 ATPase complexes were designed as an alternative treatment strategy. This resulted in significantly reduced protein levels and increased apoptosis, suggesting that targeted degradation of these complexes may be an attractive therapeutic strategy [119]. Furthermore, PROTACs have proven to act like a catalyst, working many times in a row to degrade multiple proteins. Their effect is rapid even at low concentrations, and the duration of activity is sustained over time since the cell must re-synthesize the protein of interest [120]. PROTACS taking advantage of BET inhibitors such as JQ1 and OTX015 (Birabriseb) have been evaluated in various cellular models of cancer including prostate cancer, lymphoma, and leukemia [121][111][122]. Importantly, since bromodomain inhibitors have been shown to increase the stability of BRD4, degradation has proven to be an effective method to overcome resistance [98].
5. Conclusions
References
- Mehta, V.; Trinkle-Mulcahy, L. Recent advances in large-scale protein interactome mapping. F1000Research 2016, 5.
- Bienfait, B.; Ertl, P. JSME: A free molecule editor in JavaScript. J. Cheminform. 2013, 5, 24.
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073.
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417.
- Klingbeil, O.; Lesche, R.; Gelato, K.A.; Haendler, B.; Lejeune, P. Inhibition of BET bromodomain-dependent XIAP and FLIP expression sensitizes KRAS-mutated NSCLC to pro-apoptotic agents. Cell Death Dis. 2016, 7, e2365.
- Lewin, J.; Soria, J.C.; Stathis, A.; Delord, J.P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014.
- Vazquez, R.; Riveiro, M.E.; Astorgues-Xerri, L.; Odore, E.; Rezai, K.; Erba, E.; Panini, N.; Rinaldi, A.; Kwee, I.; Beltrame, L.; et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget 2017, 8, 7598–7613.
- Odore, E.; Lokiec, F.; Cvitkovic, E.; Bekradda, M.; Herait, P.; Bourdel, F.; Kahatt, C.; Raffoux, E.; Stathis, A.; Thieblemont, C.; et al. Phase I Population Pharmacokinetic Assessment of the Oral Bromodomain Inhibitor OTX015 in Patients with Haematologic Malignancies. Clin. Pharm. 2016, 55, 397–405.
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204.
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195.
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129.
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763.
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123.
- Piha-Paul, S.A.; Hann, C.L.; French, C.A.; Cousin, S.; Brana, I.; Cassier, P.A.; Moreno, V.; de Bono, J.S.; Harward, S.D.; Ferron-Brady, G.; et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. Jnci Cancer Spectr. 2020, 4, pkz093.
- Xie, F.; Huang, M.; Lin, X.; Liu, C.; Liu, Z.; Meng, F.; Wang, C.; Huang, Q. The BET inhibitor I-BET762 inhibits pancreatic ductal adenocarcinoma cell proliferation and enhances the therapeutic effect of gemcitabine. Sci. Rep. 2018, 8, 8102.
- Seal, J.; Lamotte, Y.; Donche, F.; Bouillot, A.; Mirguet, O.; Gellibert, F.; Nicodeme, E.; Krysa, G.; Kirilovsky, J.; Beinke, S.; et al. Identification of a novel series of BET family bromodomain inhibitors: Binding mode and profile of I-BET151 (GSK1210151A). Bioorg. Med. Chem. Lett. 2012, 22, 2968–2972.
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578, 306–310.
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394.
- Aggarwal, R.R.; Schweizer, M.T.; Nanus, D.M.; Pantuck, A.J.; Heath, E.I.; Campeau, E.; Attwell, S.; Norek, K.; Snyder, M.; Bauman, L.; et al. A Phase Ib/IIa Study of the Pan-BET Inhibitor ZEN-3694 in Combination with Enzalutamide in Patients with Metastatic Castration-resistant Prostate Cancer. Clin. Cancer Res. 2020, 26, 5338–5347.
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 672–680.
- Bevill, S.M.; Olivares-Quintero, J.F.; Sciaky, N.; Golitz, B.T.; Singh, D.; Beltran, A.S.; Rashid, N.U.; Stuhlmiller, T.J.; Hale, A.; Moorman, N.J.; et al. GSK2801, a BAZ2/BRD9 Bromodomain Inhibitor, Synergizes with BET Inhibitors to Induce Apoptosis in Triple-Negative Breast Cancer. Mol. Cancer Res. 2019, 17, 1503–1518.
- Chen, P.; Chaikuad, A.; Bamborough, P.; Bantscheff, M.; Bountra, C.; Chung, C.W.; Fedorov, O.; Grandi, P.; Jung, D.; Lesniak, R.; et al. Discovery and Characterization of GSK2801, a Selective Chemical Probe for the Bromodomains BAZ2A and BAZ2B. J. Med. Chem. 2016, 59, 1410–1424.
- Zucconi, B.E.; Makofske, J.L.; Meyers, D.J.; Hwang, Y.; Wu, M.; Kuroda, M.I.; Cole, P.A. Combination Targeting of the Bromodomain and Acetyltransferase Active Site of p300/CBP. Biochemistry 2019, 58, 2133–2143.
- Picaud, S.; Fedorov, O.; Thanasopoulou, A.; Leonards, K.; Jones, K.; Meier, J.; Olzscha, H.; Monteiro, O.; Martin, S.; Philpott, M.; et al. Generation of a Selective Small Molecule Inhibitor of the CBP/p300 Bromodomain for Leukemia Therapy. Cancer Res. 2015, 75, 5106–5119.
- Palmer, W.S.; Poncet-Montange, G.; Liu, G.; Petrocchi, A.; Reyna, N.; Subramanian, G.; Theroff, J.; Yau, A.; Kost-Alimova, M.; Bardenhagen, J.P.; et al. Structure-Guided Design of IACS-9571, a Selective High-Affinity Dual TRIM24-BRPF1 Bromodomain Inhibitor. J. Med. Chem. 2016, 59, 1440–1454.
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412.
- Yao, D.; Zhang, J.; Wang, J.; Pan, D.; He, Z. Discovery of novel ATAD2 bromodomain inhibitors that trigger apoptosis and autophagy in breast cells by structure-based virtual screening. J. Enzym. Inhib. Med. Chem. 2020, 35, 713–725.
- Perez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenet. Off. J. DNA Methylation Soc. 2017, 12, 323–339.
- Cheng, X.; Huang, Z.; Long, D.; Jin, W. BET inhibitor bromosporine enhances 5-FU effect in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2020, 521, 840–845.
- Theodoulou, N.H.; Bamborough, P.; Bannister, A.J.; Becher, I.; Bit, R.A.; Che, K.H.; Chung, C.W.; Dittmann, A.; Drewes, G.; Drewry, D.H.; et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439.
- Martin, L.J.; Koegl, M.; Bader, G.; Cockcroft, X.L.; Fedorov, O.; Fiegen, D.; Gerstberger, T.; Hofmann, M.H.; Hohmann, A.F.; Kessler, D.; et al. Structure-Based Design of an in Vivo Active Selective BRD9 Inhibitor. J. Med. Chem. 2016, 59, 4462–4475.
- Lee, D.; Lee, D.Y.; Hwang, Y.S.; Seo, H.R.; Lee, S.A.; Kwon, J. The Bromodomain Inhibitor PFI-3 Sensitizes Cancer Cells to DNA Damage by Targeting SWI/SNF. Mol. Cancer Res. 2021, 19, 900–912.
- Sachchidanand; Resnick-Silverman, L.; Yan, S.; Mutjaba, S.; Liu, W.J.; Zeng, L.; Manfredi, J.J.; Zhou, M.M. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem. Biol. 2006, 13, 81–90.
- Hay, D.A.; Fedorov, O.; Martin, S.; Singleton, D.C.; Tallant, C.; Wells, C.; Picaud, S.; Philpott, M.; Monteiro, O.P.; Rogers, C.M.; et al. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J. Am. Chem. Soc. 2014, 136, 9308–9319.
- Hammitzsch, A.; Tallant, C.; Fedorov, O.; O’Mahony, A.; Brennan, P.E.; Hay, D.A.; Martinez, F.O.; Al-Mossawi, M.H.; de Wit, J.; Vecellio, M.; et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc. Natl. Acad. Sci. USA 2015, 112, 10768–10773.
- Conery, A.R.; Centore, R.C.; Neiss, A.; Keller, P.J.; Joshi, S.; Spillane, K.L.; Sandy, P.; Hatton, C.; Pardo, E.; Zawadzke, L.; et al. Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. eLife 2016, 5.
- Meier, J.C.; Tallant, C.; Fedorov, O.; Witwicka, H.; Hwang, S.Y.; van Stiphout, R.G.; Lambert, J.P.; Rogers, C.; Yapp, C.; Gerstenberger, B.S.; et al. Selective targeting of bromodomains of the bromodomain-PHD fingers family impairs osteoclast differentiation. ACS Chem. Biol. 2017, 12, 2619–2630.
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262.
- Gamsjaeger, R.; Webb, S.R.; Lamonica, J.M.; Billin, A.; Blobel, G.A.; Mackay, J.P. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol. Cell Biol. 2011, 31, 2632–2640.
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225.
- Morris, J.H.; Knudsen, G.M.; Verschueren, E.; Johnson, J.R.; Cimermancic, P.; Greninger, A.L.; Pico, A.R. Affinity purification-mass spectrometry and network analysis to understand protein-protein interactions. Nat. Protoc. 2014, 9, 2539–2554.
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810.
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887.
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539.
- Alanis-Lobato, G.; Andrade-Navarro, M.A.; Schaefer, M.H. HIPPIE v2.0: Enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res. 2017, 45, D408–D414.
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533.
- Fraschilla, I.; Jeffrey, K.L. The Speckled Protein (SP) Family: Immunity’s Chromatin Readers. Trends Immunol. 2020, 41, 572–585.
- Bouchier-Hayes, L.; Martin, S.J. CARD games in apoptosis and immunity. EMBO Rep. 2002, 3, 616–621.
- Bottomley, M.J.; Collard, M.W.; Huggenvik, J.I.; Liu, Z.; Gibson, T.J.; Sattler, M. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat. Struct. Biol. 2001, 8, 626–633.
- Waterfield, M.; Khan, I.S.; Cortez, J.T.; Fan, U.; Metzger, T.; Greer, A.; Fasano, K.; Martinez-Llordella, M.; Pollack, J.L.; Erle, D.J.; et al. The transcriptional regulator Aire coopts the repressive ATF7ip-MBD1 complex for the induction of immunotolerance. Nat. Immunol. 2014, 15, 258–265.
- Zhang, X.; Zhao, D.; Xiong, X.; He, Z.; Li, H. Multifaceted Histone H3 Methylation and Phosphorylation Readout by the Plant Homeodomain Finger of Human Nuclear Antigen Sp100C. J. Biol. Chem. 2016, 291, 12786–12798.
- Zucchelli, C.; Tamburri, S.; Filosa, G.; Ghitti, M.; Quilici, G.; Bachi, A.; Musco, G. Sp140 is a multi-SUMO-1 target and its PHD finger promotes SUMOylation of the adjacent Bromodomain. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 456–465.
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073.
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917.
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287.
- Ott, C.J.; Kopp, N.; Bird, L.; Paranal, R.M.; Qi, J.; Bowman, T.; Rodig, S.J.; Kung, A.L.; Bradner, J.E.; Weinstock, D.M. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012, 120, 2843–2852.
- Berkovits, B.D.; Wolgemuth, D.J. The role of the double bromodomain-containing BET genes during mammalian spermatogenesis. Curr. Top. Dev. Biol. 2013, 102, 293–326.
- Wu, Q.; Heidenreich, D.; Zhou, S.; Ackloo, S.; Kramer, A.; Nakka, K.; Lima-Fernandes, E.; Deblois, G.; Duan, S.; Vellanki, R.N.; et al. A chemical toolbox for the study of bromodomains and epigenetic signaling. Nat. Commun. 2019, 10, 1915.
- Hogg, S.J.; Newbold, A.; Vervoort, S.J.; Cluse, L.A.; Martin, B.P.; Gregory, G.P.; Lefebure, M.; Vidacs, E.; Tothill, R.W.; Bradner, J.E.; et al. BET Inhibition Induces Apoptosis in Aggressive B-Cell Lymphoma via Epigenetic Regulation of BCL-2 Family Members. Mol. Cancer Ther. 2016, 15, 2030–2041.
- Cummin, T.E.C.; Cox, K.L.; Murray, T.D.; Turaj, A.H.; Dunning, L.; English, V.L.; Fell, R.; Packham, G.; Ma, Y.; Powell, B.; et al. BET inhibitors synergize with venetoclax to induce apoptosis in MYC-driven lymphomas with high BCL-2 expression. Blood Adv. 2020, 4, 3316–3328.
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542.
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417.
- Yang, G.; Zhou, D.; Li, J.; Wang, W.; Zhong, W.; Fan, W.; Yu, M.; Cheng, H. VDAC1 is regulated by BRD4 and contributes to JQ1 resistance in breast cancer. Oncol. Lett. 2019, 18, 2340–2347.
- Shu, S.; Wu, H.J.; Ge, J.Y.; Zeid, R.; Harris, I.S.; Jovanovic, B.; Murphy, K.; Wang, B.; Qiu, X.; Endress, J.E.; et al. Synthetic Lethal and Resistance Interactions with BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Mol. Cell 2020.
- Esteve-Arenys, A.; Valero, J.G.; Chamorro-Jorganes, A.; Gonzalez, D.; Rodriguez, V.; Dlouhy, I.; Salaverria, I.; Campo, E.; Colomer, D.; Martinez, A.; et al. The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma. Oncogene 2018, 37, 1830–1844.
- Zhao, X.; Ren, Y.; Lawlor, M.; Shah, B.D.; Park, P.M.C.; Lwin, T.; Wang, X.; Liu, K.; Wang, M.; Gao, J.; et al. BCL2 Amplicon Loss and Transcriptional Remodeling Drives ABT-199 Resistance in B Cell Lymphoma Models. Cancer Cell 2019, 35, 752–766.e9.
- Shahbazi, J.; Liu, P.Y.; Atmadibrata, B.; Bradner, J.E.; Marshall, G.M.; Lock, R.B.; Liu, T. The Bromodomain Inhibitor JQ1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin. Cancer Res. 2016, 22, 2534–2544.
- Fiskus, W.; Sharma, S.; Qi, J.; Valenta, J.A.; Schaub, L.J.; Shah, B.; Peth, K.; Portier, B.P.; Rodriguez, M.; Devaraj, S.G.; et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol. Cancer Ther. 2014, 13, 1142–1154.
- Manzotti, G.; Ciarrocchi, A.; Sancisi, V. Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy. Cancers 2019, 11, 304.
- Saygin, C.; Carraway, H.E. Emerging therapies for acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 93.
- Anwer, F.; Gee, K.M.; Iftikhar, A.; Baig, M.; Russ, A.D.; Saeed, S.; Zar, M.A.; Razzaq, F.; Carew, J.; Nawrocki, S.; et al. Future of Personalized Therapy Targeting Aberrant Signaling Pathways in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 397–405.
- Miller, A.L.; Garcia, P.L.; Yoon, K.J. Developing effective combination therapy for pancreatic cancer: An overview. Pharm. Res. 2020, 155, 104740.
- Andrikopoulou, A.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.A.; Zagouri, F. Clinical perspectives of BET inhibition in ovarian cancer. Cell Oncol. 2021, 44, 237–249.
- Andrikopoulou, A.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.A.; Zagouri, F. The emerging role of BET inhibitors in breast cancer. Breast 2020, 53, 152–163.
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9.
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559.
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763.
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412.
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381.
- Remillard, D.; Buckley, D.L.; Paulk, J.; Brien, G.L.; Sonnett, M.; Seo, H.S.; Dastjerdi, S.; Wuhr, M.; Dhe-Paganon, S.; Armstrong, S.A.; et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew. Chem. 2017, 56, 5738–5743.
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife 2018, 7.
- Letson, C.; Padron, E. Non-canonical transcriptional consequences of BET inhibition in cancer. Pharm. Res. 2019, 150, 104508.
- Sutherell, C.L.; Tallant, C.; Monteiro, O.P.; Yapp, C.; Fuchs, J.E.; Fedorov, O.; Siejka, P.; Muller, S.; Knapp, S.; Brenton, J.D.; et al. Identification and Development of 2,3-Dihydropyrrolo[1,2-a]quinazolin-5(1H)-one Inhibitors Targeting Bromodomains within the Switch/Sucrose Nonfermenting Complex. J. Med. Chem. 2016, 59, 5095–5101.
- Lu, T.; Hu, J.C.; Lu, W.C.; Han, J.; Ding, H.; Jiang, H.; Zhang, Y.Y.; Yue, L.Y.; Chen, S.J.; Jiang, H.L.; et al. Identification of small molecule inhibitors targeting the SMARCA2 bromodomain from a high-throughput screening assay. Acta Pharm. Sin. 2018, 39, 1544–1552.
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 672–680.
- Scheepstra, M.; Hekking, K.F.W.; van Hijfte, L.; Folmer, R.H.A. Bivalent Ligands for Protein Degradation in Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 160–176.
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129.
- Wu, S.; Jiang, Y.; Hong, Y.; Chu, X.; Zhang, Z.; Tao, Y.; Fan, Z.; Bai, Z.; Li, X.; Chen, Y.; et al. BRD4 PROTAC degrader ARV-825 inhibits T-cell acute lymphoblastic leukemia by targeting ‘Undruggable’ Myc-pathway genes. Cancer Cell Int. 2021, 21, 230.
- Chew, H.K. Adjuvant therapy for breast cancer: Who should get what? West. J. Med. 2001, 174, 284–287.
- Franco, H.L.; Nagari, A.; Malladi, V.S.; Li, W.; Xi, Y.; Richardson, D.; Allton, K.L.; Tanaka, K.; Li, J.; Murakami, S.; et al. Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res. 2018, 28, 159–170.
- Raisner, R.; Bainer, R.; Haverty, P.M.; Benedetti, K.L.; Gascoigne, K.E. Super-enhancer acquisition drives oncogene expression in triple negative breast cancer. PLoS ONE 2020, 15, e0235343.
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139.



