Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simone Sidoli | + 3393 word(s) | 3393 | 2021-07-01 10:24:24 | | | |
| 2 | Vivi Li | Meta information modification | 3393 | 2021-08-04 04:19:41 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sidoli, S. Mass Spectrometry and Chromatin Compaction. Encyclopedia. Available online: https://encyclopedia.pub/entry/12724 (accessed on 26 June 2026).
Sidoli S. Mass Spectrometry and Chromatin Compaction. Encyclopedia. Available at: https://encyclopedia.pub/entry/12724. Accessed June 26, 2026.
Sidoli, Simone. "Mass Spectrometry and Chromatin Compaction" Encyclopedia, https://encyclopedia.pub/entry/12724 (accessed June 26, 2026).
Sidoli, S. (2021, August 03). Mass Spectrometry and Chromatin Compaction. In Encyclopedia. https://encyclopedia.pub/entry/12724
Sidoli, Simone. "Mass Spectrometry and Chromatin Compaction." Encyclopedia. Web. 03 August, 2021.
Copy Citation
Chromatin accessibility is a major regulator of gene expression. Histone writers/erasers have a critical role in chromatin compaction, as they “flag” chromatin regions by catalyzing/removing covalent post-translational modifications on histone proteins. Anomalous chromatin decondensation is a common phenomenon in cells experiencing aging and viral infection. Moreover, about 50% of cancers have mutations in enzymes regulating chromatin state. Numerous genomics methods have evolved to characterize chromatin state, but the analysis of (in)accessible chromatin from the protein perspective is not yet in the spotlight.
chromatin
DNA methylation
histone
mass spectrometry
post-translational modification
proteome
1. Introduction
Chromatin accessibility has a fundamental role in a wide range of biological processes including gene regulation and DNA repair. While the dogma open chromatin = transcribed genes still stands, there is still much unexplored in understanding which molecular mechanisms regulate chromatin accessibility, which are inherited, and which are failing in disease pathogenesis [1]. Numerous reviews discuss the genomics strategies and the know-how on chromatin accessibility in much detail, including an excellent recent review of Klemm et al. [2]. However, most of the data we have available come from DNA-centric approaches, i.e., high-throughput sequencing. Protein-centric studies are far less common. One exception might appear to be chromatin immunoprecipitation-sequencing (ChIP-seq) [3][4][5], as it maps protein occupancy on the chromatin; but still it provides DNA reads rather than protein data.
Proteins are actually critical players in chromatin state and dynamics. In eukaryotic cells, DNA exists in close association with histone proteins, which form nucleosomes every ≈147 bp of DNA. Canonical histones are H2A, H2B, H3, and H4, and they are bound especially in heterochromatic silenced domains by the linker histone H1 [6]. Histones are highly decorated by post-translational modifications (PTMs), which contribute to chromatin accessibility directly through their charge state or indirectly by recruiting other proteins involved in chromatin modulation (Figure 1). An example of direct accessibility is histone methylation that occurs on lysine and arginine residues. In mammalian cells, one of the best-studied marks is H3K9me2/3 (histone H3 tails di/trimethylated on the lysine residue 9), which are mainly found in constitutive heterochromatin [7]. Another example is histone acetylation, or rather acylation; acyl groups added on lysine residues neutralize the positive charge of the amino acid, which reduces the electrostatic interaction with the negatively charged DNA. For instance, acetylation of histone H3 is associated with active chromatin and plays a fundamental role in transcriptional activation [8]. Beside the abundant acetylation, histone propionylation, malonylation, crotonylation, butyrylation, succinylation, glutarylation, 2-hydroxyisobutyrylation, and β-hydroxybutyrylation are other examples of acylations detected on histone proteins [9]. Proteins with domains that bind those modifications are defined as “readers”. Some of them are transcription factors, whose duty is to recruit RNA polymerases for transcription [10]. Selected readers contribute to chromatin remodeling, i.e., rearranging DNA from a compacted to transcriptionally accessible state, or vice versa. These chromatin remodelers may directly bind DNA motifs rather than histone modifications, e.g., the SWI/SNF complex has high affinity for DNA and it is required for the enhancement of transcription by many transcriptional activators in yeast [11]. As well, other chromatin readers recognize silencing marks and contribute to chromatin compaction. An example of a protein involved in maintaining condensed heterochromatin is the Heterochromatin Protein 1 (HP1), which recognizes and binds H3K9me3 [12]. The spatial positioning of chromatin in the cell nucleus is also contributing to its accessibility [13]. For instance, Lamina-Associated Domains, or LADs, are heterochromatic domains sequestered at the nuclear periphery. Interestingly, those domains are heavily decorated by a selected histone mark, H3K9me2 [14]. As well, the nuclear pore complex associates primarily with DNA regions silenced by the Polycomb Repressive Complex, at least in Drosophila [15]. In summary, the fine-tuning of protein–DNA interactions, protein–protein interactions, and protein modifications (especially histones) are critical contributors to chromatin accessibility. Intuitively, mutations and anomalous protein regulations can have a dramatic effect on the cell phenotype.
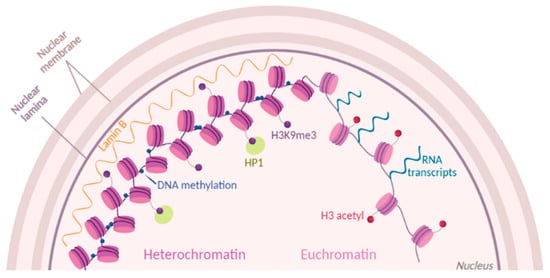
Figure 1. Chromatin dynamics. Chromatin state is modulated by histone post-translational modifications (PTMs) (purple and red dots). Part of condensed heterochromatin is located at the nuclear periphery and is enriched in methylations of histone H3 at the residue K9 (H3K9me3). Readers of this modifications like Lamin B and HP1 maintain the chromatin in a compacted state. Euchromatin is shown as open and accessible chromatin, enriched for histone acetylation (H3 acetyl) and prone to transcription.
Events like UV exposure, smoking, viral infection, cancer, and aging correlate with chromatin decondensation, i.e., large and small heterochromatic domains become euchromatic (accessible and prone to transcription) [16]. In fact, chromatin decondensation is not only an issue in terms of uncontrolled gene expression; chromatin domains decorated by H3K9me2/3 are rich in DNA repetitive units such as transposons, ALUs, and other satellite regions [17], and their accessibility to the transcriptional machinery is harmful for the cell [18]. Together, DNA methylation, histone PTMs, and non-coding regions ensure proper chromatin conformation and promote genome stability [19].
One of the main features of cancer is the (epi)genome instability. The transition from normal tissue to cancer is sometimes characterized by changes in the distribution of H3K9me2/3, in HP1 expression levels [12] and by regional loss of heterochromatin which, in turn, become euchromatin [16]. Additionally, DNA hypomethylation of CpG dinucleotides in the pericentromeric region of the chromosome might be involved in many types of tumors [20]. All these changes directly affect the transcriptional activity and genomic stability, leading to cellular uncontrolled proliferation and metastasis. Interestingly, elevated levels of methylation, especially in gene promoter regions, is related to aberrant silencing of transcription and inactivation of tumor-suppressor genes [20].
Reduced global heterochromatin, altered histone marks, and global hypomethylation of DNA have also been associated with aging. Significant changes in global nuclear architecture during physiological aging, as well as altered gene expression, might be triggered by the loss of heterochromatin domains [21]. This global loss was observed in human old fibroblasts and fibroblasts from Hutchinson–Gilford progeria syndrome (HGPS), indicating that several components are shared between normal aging and accelerated aging syndromes [22]. Senescent cells, during aging, present different levels of histone variants. An example is the loss of canonical histone H3.1 and H3.2 and the increase of the histone variant H3.3, which is incorporated into the genome in a replication-independent manner and plays a key role in chromatin maintenance when cells are no longer dividing [21]. MacroH2A is another histone variant that promotes transcriptional silencing and is abundant in SAHF (senescence associated heterochromatin foci), in addition to being a critical regulator of chromatin dynamics during senescence [21][23]. In fact, chromatin structure is under dynamic changes throughout the entire life span of an organism. Among the histone modifications that are known to affect the longevity process, the most important ones are acetylation and methylation of lysine residues. Increased levels of H4K16ac lead to more open chromatin and, in old yeast cells, it correlates with decreased silencing of reporter genes and shortened lifespan. Conversely, reduced levels of H4K16ac is beneficial for longevity in yeast, due to a more closed global chromatin structure [24].
External events also affect chromatin state. DNA damage accumulates with age, but the process can be accelerated by reactive oxygen species (ROS), exposure to UV irradiation, and alcohol intake. Oxidative stress occurs because of ROS accumulation, affecting chromatin and chromatin modifying-enzymes. In general, it can stimulate global heterochromatin loss and modify histones folding and stability, as well as their PTMs, influencing the expression of genes that are normally in a silenced state [25][26][27]. Besides oxidative stress effects, exposure to ionizing radiation leads to less compact heterochromatin, which adopts a more loose structure [28]. At the same time, there is evidence that radiation induces global compaction of chromatin, indicating a potential mechanism to protect genome integrity [29][30]. Metabolites generated during ethanol metabolism can also impact chromatin structure. Animal experiments demonstrated that excessive alcohol intake modifies the mechanisms regulating chromatin remodeling and gene expression by altering the levels of histone acetylation as well as DNA methylation [31].
2. Mass Spectrometry to Study Chromatin State: First Steps with Nucleotide Modifications
DNA methylation is a known marker of DNA silencing which regulates gene expression and epigenetics inheritance [32][33]. Chromatin domains with methylated DNA are associated with compacted heterochromatin states. This prompted the need for genome-wide DNA methylation analysis and resulted in the continuous evolution of various analytical methods involving bisulfite reactions, the use of methylation-sensitive restriction enzymes, radiolabeling, immunoassays, methylation specific PCR, microarray technology, next generation sequencing, thin layer chromatography (TLC), and reversed phase high pressure liquid chromatography (RP-HPLC) and with mass spectrometry [34]. However, the two most popular methods remain ELISA and mass spectrometry [35]. Immunoassays are arguably faster and simpler, but they tend to be more variable due to non-specificity issues. Mass spectrometry is considered as the “gold standard” due to the high sensitivity and specificity of the technique, but it is not as robust and straightforward.
Paper, thin layer, ion exchange and gas chromatography coupled to either UV or mass spectrometry are historical methods to separate and quantify the four major DNA bases (G, C, A, T) and the methylated DNA bases (5mdC and 5hmdC). In 1980, Kuo and colleagues successfully used C18 chromatography coupled to UV detection to quantify 1–2% 5mdC from DNA of calf thymus and salmon sperm [36]. DNA was previously digested into individual nucleosides using DNase I, nuclease P1, and alkaline phosphatase. Another study reported the use of electrophoretic derivatization and electron-capture negative chemical ionization combined with moving belt liquid chromatography-mass spectrometry to quantify 5mdC and 5hmdC [37]. The method has then evolved including a combination of (1) DNA hydrolysis using HpaII and MspI restriction nucleases, (2) electron ionization gas chromatography, (3) C18 chromatography, and (4) hybridization analysis using a series of probes. Together, this was used to map the methylated regions of DNA containing an actively and differentially expressed somatic H1 histone gene from sperm, embryo, and adult tissues of Chaetopterus worm [38].
DNA methylation was quantified in disease states like leukemia using urine samples [39]. Chromatographic separation has required optimization, mostly because small molecules like nucleosides have weak hydrophobic interaction with reversed-phase C18 columns. Further optimization included varying methanol solvent to decrease surface tension, addition of acetic acid for protonation and found that ammonium acetate/methanol (88:12 v/v) is the best for both chromatographic separation and detection in mass spectrometry using electrospray ionization [39]. The addition of two different RNAses and re-precipitation of DNA was importantly discussed to at least minimize possible interference from 5-methylcytidine residues from tRNA and rRNA contaminants [40]. Song and colleagues [41] reported a chromatographic separation for the efficient separation and detection of 5mdC and 5hmdC from the other four deoxyribonucleosides and methylated RNA nucleoside contaminants by electrospray ionization tandem mass spectrometry using a triple quadrupole mass spectrometer from genomic DNA. Methylated DNA was also analyzed using mass spectrometry in embryonic stem cells by measuring 5hmdC and 5mdC [42].
Ito and colleagues discovered that 5mC is not only converted to 5hmC but also into 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) by TET proteins [43]. Those modified nucleosides represented a greater challenge for quantification due to their lower abundance; 1–20 × 106 cytosine for 5fC and 3 × 106 cytosine for 5caC. In fact, they required modifications to the chromatographic setup to achieve sufficient sensitivity [35]. Chemical derivatization using 2-bromo-1-(4-dimethylamino-phenyl)-ethanone (BDAPE) was introduced to selectively label 5mdC, 5hmdC, 5fdC, and 5cadC [44]. This enhanced sensitivity of 35–123-fold compared to un-derivatized cytosine modifications [35][44]. More recently, hydrophilic interaction liquid chromatography (HILIC) [35][45] and porous graphitic carbon (PGC) [46] have emerged as potential alternatives to C18-based chromatography for nucleosides. However, a robust platform for nucleoside quantification using mass spectrometry is still less common in research labs than one could expect. In summary, quantification of DNA modifications has been the pioneer of chromatin state analysis using mass spectrometry since its establishment about 40 years ago. However, for a comprehensive overview of molecular components of accessible and inaccessible chromatin, a protein perspective is required.
3. Multi-Dimensional Histone Modification Analysis Using Mass Spectrometry
Mass spectrometry is currently the only technique that can identify and quantify in a large-scale manner the relative abundance of histone PTMs [47]. Other techniques, such as Western blotting, ELISA, or immunohistochemistry, may be used for histone PTM quantification. However, all of these methods rely on specific antibodies, which may not be readily available for some PTMs or may provide inaccurate quantitation due to cross-reactivity or epitope masking [48][49]. Mass spectrometry is used to quantify both single and combinatorial histone PTMs [50], although it is important to note that extracted histones are decoupled with their original DNA location and thus this analysis does not allow to define the genome-wide distribution of histone marks. Creative approaches have been exploited in mass spectrometry to look beyond the sole abundance of histone PTMs; e.g., the group of Anja Groth used metabolic labeling in cell culture to monitor whether newly synthesized or recycled histones were transferred into the newly replicated DNA [51]. Nevertheless, this approach cannot define the location of these histones nor provide direct information about their accessibility.
The traditional peptide centric analysis of histone proteins (bottom-up) is similar to the typical proteomics pipeline. Specifically, proteins are usually digested with the protease trypsin (cleaves after lysine and arginine residues) into short (4–20 aa) peptides for analysis, as both chromatographic separation and detection by mass spectrometry are more robust and sensitive than intact protein analysis (top-down). However, histones are very basic proteins, i.e., rich in arginine and lysine residues, and thus trypsin digestion would result in excessively short peptides for reconstructing their position on the protein. For this reason, derivatization of lysine residues is frequently applied to modify the side chain of lysine residues so that trypsin can only cleave arginine residues and generate proper size peptides [47]. Since 2004, the sample preparation strategy has been periodically optimized and different laboratories have applied different derivatization methods to generate proper size peptides, i.e., from the use of D6-acetic anhydride [52], to propionic anhydride [53] to NHS-propionate [54] to phenyl isocyanate [8]. Independently from the protocol applied, it is now clear that by performing chemical labeling of lysine residues assists more confident and accurate PTM quantification. An overview of the different sample preparation strategies, including their advantages and disadvantages, is described elsewhere [55][56][57][58][59].
While histone PTM quantification per se does not provide direct information about chromatin state, the relative abundance of selected histone marks is used to define how accessible chromatin is overall. For instance, hyperacetylation of histone H4 on the residues K5/K8/K12/K16 (in particular K16 [60]) reveals that chromatin is relatively unfolded. As well, the increase in abundance of selected silencing marks has been interpreted as “restricted” chromatin environment, e.g., in schizophrenia [61]. Those are indirect conclusions to the chromatin state, and we should not forget that hundreds of histone marks have been identified but still not assigned to either accessible or inaccessible chromatin. The advent of “middle-down” mass spectrometry showed an even more complicated picture; this strategy is named as such as it is a compromise between the peptide-based approach (bottom-up) and the analysis of intact undigested proteins (top-down). Middle-down utilizes proteases that cleave rare amino acid residues on histone sequences, i.e., aspartic and glutamic acid, to generate intact histone N-terminal tails (50–60 aa) [62]. Identifying and quantifying these long polypeptides is equivalent to mapping co-existing PTMs on the same histone protein, i.e., this approach can be used to define combinatorial PTM codes [62]. Notably, other strategies not based on mass spectrometry have been implemented to study combinatorial modifications; Shema and co-workers developed an antibody-based imaging platform to map co-existing modifications on nucleosomes [63], while Sadeh and colleagues established a method named Combinatorial-iChIP to map genome-wide the co-occurrence of two histone PTMs instead of the typical single PTM analysis of canonical ChIP-seq [64]. These methods offer undeniable advantages; on the other hand, middle-down mass spectrometry is independent from antibodies and it is not limited in the number of co-existing modifications to quantify on a single polypeptide. From middle-down data, it became rapidly clear that histones are very rarely decorated with one or two modifications in the cells, but they rather have 5–8 co-existing marks on the same histone protein [65]. Frequently, those PTMs have unknown biological function or presumed opposite roles on chromatin. Why do they co-exist then? This is still an unanswered question, as there is currently no technology that can define the accessibility on chromatin of hypermodified histone codes.
The differential turnover of nucleosomes has been described in multiple publications [66][67][68], firmly establishing that nucleosomes are exchanged from chromatin multiple times within a cell cycle. This opens an opportunity for mass spectrometry, as protein turnover can be quantified by metabolic labeling (e.g., Zee et al. [69]). In a recent work, we have assessed that metabolic labeling of histones can be utilized to define whether a certain modification is on actively transcribed chromatin or inaccessible [70]. The principle is based on cell cultures feeding on stable isotope labeled amino acids, which are partially incorporated in the histone amino acid sequence (Figure 2). Those histones with higher recycling rates will have a relatively higher heavy/light ratio, and this recycling rate is more frequent on chromatin domains with active transcription. Interestingly, this labeling has the potential to be utilized for middle-down [71], paving the way to the determination of the accessibility on chromatin of combinatorial histone codes. However, part of the current challenge is developing the proper bioinformatics to discriminate signals corresponding to combinatorial modifications vs. partial metabolic labeling.
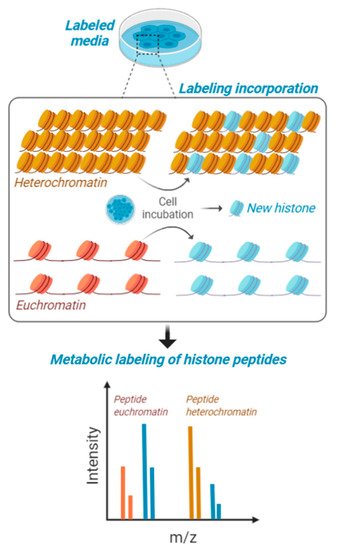
Figure 2. Metabolic labeling of histones peptides. Cells in culture are fed with media containing stable isotope labeled amino acids and are maintained for a certain interval of time to produce about 50% of newly synthesized histones. This interval of time is contingent to their proliferation rate, as the population needs to undergo at least one cell cycle for proper labeling. Heavy amino acids are incorporated in the histone amino acid sequence and then cells are processed for histone PTM analysis. Accessible chromatin (euchromatin—in orange) is labeled with higher rate compared to condensed heterochromatin (in yellow). Isotopic labeling is represented in blue.
4. Quantifying the Chromatin-State Dependent Proteome with Mass Spectrometry
Proteomics has become a discipline with many applications, most of them contingent with appropriate sample preparation. The routine procedure of sample preparation for proteomics is arguably one of the simplest among the -omics; most extracted or purified proteins are soluble in water, and they can be prepared for mass spectrometry with a rapid three steps procedure, i.e., reduction, alkylation, and digestion into peptides. For this reason, a myriad of alternative procedures have been engineered to enhance sensitivity, specificity, and quantitative dimensions (time, localization, interactions, turnover rate). In other words, we can analyze the proteome of a specific chromatin state if we establish a dedicated chromatin fractionation procedure that allows to physically isolate chromatin fractions and analyze them separately by mass spectrometry or selectively label those domains (Figure 3). In 2003, Shiio and colleagues, in a pioneering study, developed a method to identify and quantify chromatin-associated regulatory factors by a combination of chromatin isolation and mass spectrometry analysis [72]. A recent approach was named “gradient-seq”, a method in which chromatin is cross-linked and afterwards fractionated over a sucrose gradient based on its resistance to sonication [73]. Cross-linked heterochromatic domains generate larger macromolecular structures, which can be separated by smaller accessible euchromatic domains. However, this method is unable to define the histone PTMs associated with more subtle differences in chromatin compaction since it is largely limited to resolving heterochromatin from euchromatin. Hybridization capture of chromatin-associated proteins for proteomics (HyCCAPP) is an approach developed to identify the protein components of alphoid chromatin, which is rich in a highly repetitive class of DNA [74]. Using this method coupled to mass spectrometry, Buxton and colleagues were able to analyze human protein–alpha satellite interactions. Moreover, locus specific proteomics was performed by exploiting the pull-down of a specific DNA region; two protocols named proteomics of isolated chromatin segments (PICh) [75] and insertional chromatin immunoprecipitation (iChIP) [76] were optimized for the direct identification of the bound proteome. PICh is based on nucleic acid probes, while iChIP utilizes antibodies to precipitate specific proteins benchmarking the locus of interest, e.g., CTCF was targeted to identify insulator complexes, which function as boundaries of chromatin domains. Alternatively, synthetic chromatin was also engineered with histone PTMs using ligation to purify proteins binding to selected histone marks [77].
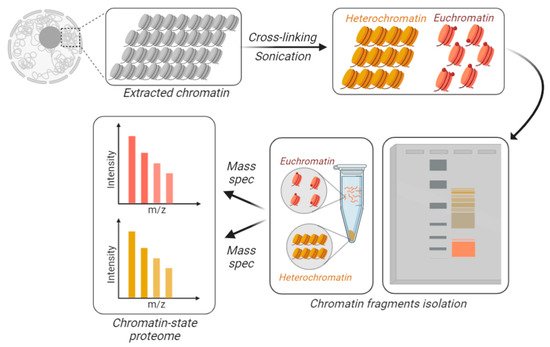
Figure 3. Chromatin-state proteome analysis. To differentially identify and quantify proteins from accessible vs. inaccessible chromatin, the chromatin is physically fractionated into separate tubes. DNA is cross-linked and the extracted chromatin is fractionated based on its resistance to sonication. Larger macromolecules are the result of sonicated heterochromatin (in yellow), while accessible chromatin is sheared into smaller fractions, i.e., euchromatin (in orange). Those fractions can be separated using gels or centrifugation.
References
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J.P. Targeting Epigenetic Readers in Cancer. N. Engl. J. Med. 2012, 367, 647–657.
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220.
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837.
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 2007, 316, 1497–1502.
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560.
- He, S.; Vickers, M.; Zhang, J.; Feng, X. Natural depletion of histone H1 in sex cells causes DNA demethylation, heterochromatin decondensation and transposon activation. eLife 2019, 8, 8.
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705.
- Maile, T.M.; Izrael-Tomasevic, A.; Cheung, T.; Guler, G.D.; Tindell, C.; Masselot, A.; Liang, J.; Zhao, F.; Trojer, P.; Classon, M.; et al. Mass spectrometric quantification of histone post-translational modifications by a hybrid chemical labeling method. Mol. Cell. Proteom. 2015, 14, 1148–1158.
- Simithy, J.; Sidoli, S.; Yuan, Z.-F.; Coradin, M.; Bhanu, N.V.; Marchione, D.M.; Klein, B.J.; Bazilevsky, G.A.; McCullough, C.E.; Magin, R.; et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 2017, 8, 1141.
- Bueno, R.R.; Ruiz, P.D.L.C.; Artal-Sanz, M.; Askjaer, P.; Dobrzynska, A. Nuclear Organization in Stress and Aging. Cells 2019, 8, 664.
- Quinn, J.; Fyrberg, A.M.; Ganster, R.W.; Schmidt, M.C.; Peterson, C.L. DNA-binding properties of the yeast SWI/SNF complex. Nature 1996, 379, 844–847.
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Boil. 2018, 34, 265–288.
- Harr, J.; Luperchio, T.R.; Wong, X.; Cohen, E.; Wheelan, S.J.; Reddy, K. Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. J. Cell Boil. 2015, 208, 33–52.
- Poleshko, A.; Smith, C.L.; Nguyen, S.C.; Sivaramakrishnan, P.; Wong, K.G.; Murray, J.I.; Lakadamyali, M.; Joyce, E.F.; Jain, R.; Epstein, J.A.; et al. H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis. eLife 2019, 8, 8.
- Gozalo, A.; Duke, A.; Lan, Y.; Pascual-Garcia, P.; Talamas, J.A.; Nguyen, S.C.; Shah, P.P.; Jain, R.; Joyce, E.F.; Capelson, M. Core Components of the Nuclear Pore Bind Distinct States of Chromatin and Contribute to Polycomb Repression. Mol. Cell 2020, 77, 67–81.
- Feinberg, A.P. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N. Engl. J. Med. 2018, 378, 1323–1334.
- Bartlett, A.A.; Hunter, R.G. Transposons, stress and the functions of the deep genome. Front. Neuroendocr. 2018, 49, 170–174.
- McMurchy, A.N.; Stempor, P.; Gaarenstroom, T.; Wysolmerski, B.; Dong, Y.; Aussianikava, D.; Appert, A.; Huang, N.; Kolasinska-Zwierz, P.; Sapetschnig, A.; et al. A team of heterochromatin factors collaborates with small RNA pathways to combat repetitive elements and germline stress. eLife 2017, 6.
- Feng, J.X.; Riddle, N.C. Epigenetics and genome stability. Mamm. Genome 2020, 1–15.
- Herman, J.G.; Baylin, S.B. Gene Silencing in Cancer in Association with Promoter Hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054.
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584.
- Feser, J.; Tyler, J.K. Chromatin structure as a mediator of aging. FEBS Lett. 2010, 585, 2041–2048.
- Scaffidi, P.; Misteli, T. Lamin A-Dependent Nuclear Defects in Human Aging. Science 2006, 312, 1059–1063.
- Kane, A.E.; Sinclair, D.A. Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Boil. 2019, 54, 61–83.
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862.
- Niu, Y.; Desmarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free. Radic. Boil. Med. 2015, 82, 22–28.
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Boil. 2013, 1, 8–16.
- Zhang, Y.; Máté, G.; Müller, P.; Hillebrandt, S.; Krufczik, M.; Bach, M.; Kaufmann, R.; Hausmann, M.; Heermann, D.W. Radiation Induced Chromatin Conformation Changes Analysed by Fluorescent Localization Microscopy, Statistical Physics, and Graph Theory. PLoS ONE 2015, 10, e0128555.
- Schick, S.; Fournier, D.; Thakurela, S.; Sahu, S.K.; Garding, A.; Tiwari, V.K. Dynamics of chromatin accessibility and epigenetic state in response to UV damage. J. Cell Sci. 2015, 128, 4380–4394.
- Hamilton, C.; Hayward, R.L.; Gilbert, N. Global chromatin fibre compaction in response to DNA damage. Biochem. Biophys. Res. Commun. 2011, 414, 820–825.
- Warnault, V.; Darcq, E.; Levine, A.; Barak, S.; Ron, D. Chromatin remodeling—A novel strategy to control excessive alcohol drinking. Transl. Psychiatry 2013, 3, e231.
- Holliday, R.; Pugh, J. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232.
- Riggs, A. X inactivation, differentiation, and DNA methylation. Cytogenet. Genome Res. 1975, 14, 9–25.
- Harrison, A.; Parle-McDermott, A. DNA Methylation: A Timeline of Methods and Applications. Front. Genet. 2011, 2, 74.
- Chowdhury, B.; Cho, I.-H.; Irudayaraj, J. Technical advances in global DNA methylation analysis in human cancers. J. Boil. Eng. 2017, 11, 10.
- Kuo, K.C.; McCune, R.A.; Gehrke, C.W.; Midgett, R.; Ehrlich, M. Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA. Nucleic Acids Res. 1980, 8, 4763–4776.
- Annan, R.S.; Kresbach, G.M.; Giese, R.W.; Vouros, P. Trace detection of modified dna bases via moving-belt liquid chromatography—Mass spectrometry using electrophoric derivatization and negative chemica. J. Chromatogr. A 1989, 465, 285–296.
- Del Gaudio, R.; Di Giaimo, R.; Geraci, G. Genome methylation of the marine annelid worm Chaetopterus variopedatus: Methylation of a CpG in an expressed H1 histone gene. FEBS Lett. 1997, 417, 48–52.
- Zambonin, C.G.; Palmisano, F. Electrospray ionization mass spectrometry of 5-methyl-2’-deoxycytidine and its determination in urine by liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid. Commun. Mass. Spectrom. 1999, 13, 2160–2165.
- Friso, S.; Choi, S.-W.; Dolnikowski, G.; Selhub, J. A Method to Assess Genomic DNA Methylation Using High-Performance Liquid Chromatography/Electrospray Ionization Mass Spectrometry. Anal. Chem. 2002, 74, 4526–4531.
- Song, L.; James, S.R.; Kazim, L.; Karpf, A.R. Specific Method for the Determination of Genomic DNA Methylation by Liquid Chromatography-Electrospray Ionization Tandem Mass Spectrometry. Anal. Chem. 2005, 77, 504–510.
- Le, T.; Kim, K.-P.; Fan, G.; Faull, K.F. A sensitive mass spectrometry method for simultaneous quantification of DNA methylation and hydroxymethylation levels in biological samples. Anal. Biochem. 2011, 412, 203–209.
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303.
- Tang, Y.; Zheng, S.-J.; Qi, C.-B.; Feng, Y.-Q.; Yuan, B.-F. Sensitive and Simultaneous Determination of 5-Methylcytosine and Its Oxidation Products in Genomic DNA by Chemical Derivatization Coupled with Liquid Chromatography-Tandem Mass Spectrometry Analysis. Anal. Chem. 2015, 87, 3445–3452.
- Zhang, L.; Li, Z.; Chen, G.; Huang, Q.; Zhang, J.; Wen, J.; Ye, X.; Cai, C. Validation and quantification of genomic 5-carboxylcytosine (5caC) in mouse brain tissue by liquid chromatography-tandem mass spectrometry. Anal. Methods 2016, 8, 5812–5817.
- Giessing, A.M.B.; Scott, L.G.; Kirpekar, F. A Nano-Chip-LC/MS n Based Strategy for Characterization of Modified Nucleosides Using Reduced Porous Graphitic Carbon as a Stationary Phase. J. Am. Soc. Mass Spectrom. 2011, 22, 1242–1251.
- Sidoli, S.; Bhanu, N.V.; Karch, K.; Wang, X.; Garcia, B.A. Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J. Vis. Exp. 2016, e54112.
- Egelhofer, T.A.; Minoda, A.; Klugman, S.; Lee, K.; Kolasinska-Zwierz, P.; Alekseyenko, A.A.; Cheung, M.-S.; Day, D.S.; Gadel, S.; Gorchakov, A.A.; et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Boil. 2010, 18, 91–93.
- Rothbart, S.B.; Dickson, B.M.; Raab, J.R.; Grzybowski, A.; Krajewski, K.; Guo, A.H.; Shanle, E.K.; Josefowicz, S.Z.; Fuchs, S.M.; Allis, C.D.; et al. An interactive database for the assessment of histone antibody specificity. Mol. Cell 2015, 59, 502–511.
- Sidoli, S.; Cheng, L.; Jensen, O.N. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J. Proteom. 2012, 75, 3419–3433.
- Alabert, C.; Barth, T.K.; Reverón-Gómez, N.; Sidoli, S.; Schmidt, A.; Jensen, O.N.; Imhof, A.; Groth, A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genome Res. 2015, 29, 585–590.
- Bonaldi, T.; Imhof, A.; Regula, J.T. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics 2004, 4, 1382–1396.
- Garcia, B.A.; Mollah, S.; Ueberheide, B.; Busby, S.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, N.F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007, 2, 933–938.
- Liao, R.; Wu, H.; Deng, H.; Yu, Y.; Hu, M.; Zhai, H.; Yang, P.; Zhou, S.; Yi, W. Specific and Efficient N-Propionylation of Histones with Propionic Acid N-Hydroxysuccinimide Ester for Histone Marks Characterization by LC-MS. Anal. Chem. 2013, 85, 2253–2259.
- Sidoli, S.; Yuan, Z.-F.; Lin, S.; Karch, K.; Wang, X.; Bhanu, N.; Arnaudo, A.M.; Britton, L.-M.; Cao, X.-J.; Gonzales-Cope, M.; et al. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics 2015, 15, 1459–1469.
- Soldi, M.; Cuomo, A.; Bonaldi, T. Quantitative assessment of chemical artefacts produced by propionylation of histones prior to mass spectrometry analysis. Proteomics 2016, 16, 1952–1954.
- Meert, P.; Dierickx, S.; Govaert, E.; De Clerck, L.; Willems, S.; Dhaenens, M.; Deforce, D. Tackling aspecific side reactions during histone propionylation: The promise of reversing overpropionylation. Proteomics 2016, 16, 1970–1974.
- Paternoster, V.; Edhager, A.V.; Sibbersen, C.; Nielsen, A.L.; Børglum, A.D.; Christensen, J.H.; Palmfeldt, J. Quantitative assessment of methyl-esterification and other side reactions in a standard propionylation protocol for detection of histone modifications. Proteomics 2016, 16, 2059–2063.
- Meert, P.; Govaert, E.; Scheerlinck, E.; Dhaenens, M.; Deforce, D. Pitfalls in histone propionylation during bottom-up mass spectrometry analysis. Proteomics 2015, 15, 2966–2971.
- Zhang, R.; Erler, J.; Langowski, J. Histone Acetylation Regulates Chromatin Accessibility: Role of H4K16 in Inter-nucleosome Interaction. Biophys. J. 2016, 112, 450–459.
- Chase, K.A.; Gavin, D.P.; Guidotti, A.; Sharma, R.P. Histone methylation at H3K9: Evidence for a restrictive epigenome in schizophrenia. Schizophr. Res. 2013, 149, 15–20.
- Sidoli, S.; Garcia, B.A. Middle-down proteomics: A still unexploited resource for chromatin biology. Expert Rev. Proteom. 2017, 14, 617–626.
- Shema, E.; Jones, D.; Shoresh, N.; Donohue, L.; Ram, O.; Bernstein, B.E. Single-molecule decoding of combinatorially modified nucleosomes. Science 2016, 352, 717–721.
- Sadeh, R.; Launer-Wachs, R.; Wandel, H.; Rahat, A.; Friedman, N. Elucidating Combinatorial Chromatin States at Single-Nucleosome Resolution. Mol. Cell 2016, 63, 1080–1088.
- Sidoli, S.; Schwämmle, V.; Ruminowicz, C.; Hansen, T.A.; Wu, X.; Helin, K.; Jensen, O.N. Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics 2014, 14, 2200–2211.
- Deal, R.B.; Henikoff, J.G.; Henikoff, S. Genome-Wide Kinetics of Nucleosome Turnover Determined by Metabolic Labeling of Histones. Science 2010, 328, 1161–1164.
- Dion, M.; Kaplan, T.; Kim, M.; Buratowski, S.; Friedman, N.; Rando, O.J. Dynamics of Replication-Independent Histone Turnover in Budding Yeast. Science 2007, 315, 1405–1408.
- Radman-Livaja, M.; Verzijlbergen, K.F.; Weiner, A.; Van Welsem, T.; Friedman, N.; Rando, O.J.; Van Leeuwen, F. Patterns and Mechanisms of Ancestral Histone Protein Inheritance in Budding Yeast. PLoS Boil. 2011, 9, e1001075.
- Zee, B.M.; Levin, R.S.; DiMaggio, P.A.; Garcia, B.A. Global turnover of histone post-translational modifications and variants in human cells. Epigenetics Chromatin 2010, 3, 22.
- Sidoli, S.; Lopes, M.; Lund, P.J.; Goldman, N.; Fasolino, M.; Coradin, M.; Kulej, K.; Bhanu, N.V.; Vahedi, G.; Garcia, B.A. A mass spectrometry-based assay using metabolic labeling to rapidly monitor chromatin accessibility of modified histone proteins. Sci. Rep. 2019, 9, 13613–13615.
- Sidoli, S.; Lu, C.; Coradin, M.; Wang, X.; Karch, K.; Ruminowicz, C.; Garcia, B.A. Metabolic labeling in middle-down proteomics allows for investigation of the dynamics of the histone code. Epigenetics Chromatin 2017, 10, 34.
- Shiio, Y.; Eisenman, R.N.; Yi, E.C.; Donohoe, S.; Goodlett, D.R.; Aebersold, R. Quantitative proteomic analysis of chromatin-associated factors. J. Am. Soc. Mass Spectrom. 2003, 14, 696–703.
- Becker, J.S.; McCarthy, R.L.; Sidoli, S.; Donahue, G.; Kaeding, K.; He, Z.; Lin, S.; Garcia, B.A.; Zaret, K.S. Genomic and Proteomic Resolution of Heterochromatin and its Restriction of Alternate Fate Genes. Mol. Cell 2017, 68, 1023–1037.e15.
- Buxton, K.E.; Kennedy-Darling, J.; Shortreed, M.R.; Zaidan, N.Z.; Olivier, M.; Scalf, M.; Sridharan, R.; Smith, L.M. Elucidating Protein–DNA Interactions in Human Alphoid Chromatin via Hybridization Capture and Mass Spectrometry. J. Proteome Res. 2017, 16, 3433–3442.
- Dejardin, J.; Kingston, R.E. Purification of Proteins Associated with Specific Genomic Loci. Cell 2009, 136, 175–186.
- Fujita, T.; Fujii, H. Direct Identification of Insulator Components by Insertional Chromatin Immunoprecipitation. PLoS ONE 2011, 6, e26109.
- Nikolov, M.; Stützer, A.; Mosch, K.; Krasauskas, A.; Soeroes, S.; Stark, H.; Urlaub, H.; Fischle, W. Chromatin Affinity Purification and Quantitative Mass Spectrometry Defining the Interactome of Histone Modification Patterns. Mol. Cell. Proteom. 2011, 10.
More
Information
Subjects:
Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
986
Revisions:
2 times
(View History)
Update Date:
04 Aug 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No