Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jörg Meurer | + 4272 word(s) | 4272 | 2021-08-02 10:32:49 | | | |
| 2 | Camila Xu | -9 word(s) | 4263 | 2021-08-03 11:48:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Meurer, J. The Chloroplast Epitranscriptome. Encyclopedia. Available online: https://encyclopedia.pub/entry/12702 (accessed on 26 July 2026).
Meurer J. The Chloroplast Epitranscriptome. Encyclopedia. Available at: https://encyclopedia.pub/entry/12702. Accessed July 26, 2026.
Meurer, Jörg. "The Chloroplast Epitranscriptome" Encyclopedia, https://encyclopedia.pub/entry/12702 (accessed July 26, 2026).
Meurer, J. (2021, August 03). The Chloroplast Epitranscriptome. In Encyclopedia. https://encyclopedia.pub/entry/12702
Meurer, Jörg. "The Chloroplast Epitranscriptome." Encyclopedia. Web. 03 August, 2021.
Copy Citation
Here, we report about epitranscriptomic methods for the identification of RNA modifications, bioinformatic tools, and the potential physiological roles of RNA modifiers and interpreters in plant nuclear/cytoplasmic gene expression related to chloroplast functions and the post-transcriptional fate of chloroplast RNAs.
chloroplast
RNA metabolism
epitranscriptome
RNA methylation
posttranscriptional regulation
development
acclimation
stress response
1. Introduction
The chloroplast is the result of an endosymbiotic event in which a cyanobacterium was ingested by a eukaryotic host cell. Although chloroplast biogenesis requires regulation of transcription rates, umpteen posttranscriptional events predominate in the development- and environment-dependent control of gene expression [1][2][3][4][5]. Chloroplasts have retained parts of the cyanobacterial-derived translation and RNA degradation system, such as masking of RNAs by polyadenylation, but only few exoribonucleases with little sequence specificity act on chloroplast RNA species [6]. Unlike in cyanobacteria, in plant chloroplasts nearly all polycistronic precursor transcripts are processed by endonucleases and resulting products are further stabilized and subjected to additional enzyme-based modifications [3][7][8][9][10]. This necessitated the recruitment of often plant-specific and nucleus-encoded proteins, which enable regulation of chloroplast gene expression on single gene levels [2][4][11]. Little is known about factors that authentically regulate plastid mRNA stability and/or translation and even less about the posttranscriptional control meditated by metabolic processes or endogenous and external stimuli [2][11][12][13]. The role of the plant epitranscriptome in overcoming the challenges of plant life has only just begun to be studied in depth in the different genetic compartments.
Recently, methodological advances in next-generation sequencing including nanopore sequencing, CLIP-technologies, antibody-based approaches, mass spectrometry, ribosomal profiling, as well as the use of chemical and enzymatic modifications have significantly increased our knowledge about epitranscriptomics and laid the foundation for understanding its role in regulation of gene expression in prokaryotes and eukaryotes [14].
More than 170 different modifications in coding and non-coding RNAs as well as several hundred factors involved in epitranscriptomics have been discovered [15][16][17]. The majority of the modifications are found in the bases of predominantly non-coding and coding RNAs and only a few in the phosphate or sugar backbone. While our knowledge about RNA modifications in the plant nuclear/cytoplasmic system is considerable rich, little attention has been paid to the chloroplast epitranscriptome so far [16][18][19][20][21]. As in other systems, chloroplast transcripts potentially provide multiple platforms for numerous RNA modifiers (writers and erasers) and interpreters (readers). The latter recognize the modifications presumably reflecting a complex interplay between epitranscriptome players, RNA processing, and translation and thus important parts of the chloroplast metabolism and photosynthesis. Studying the response of these readers to endogenous and external stimuli in terms of RNA binding and the readout of modifications will significantly contribute to our understanding of the coordination of plastid gene expression and metabolism beyond the mere change in RNA levels. This is especially relevant since chloroplasts are believed to function as central sensors that perceive environmental signals in order to trigger plant gene expression and most likely the organellar epitranscriptome [22].
While the role of polyadenylation in RNA degradation and mostly C to U editing events in chloroplasts are well understood [23], little is known about the nature, dynamics, and functions of other modifications such as the diverse methylation steps m6A, m1A, m7G, m5C or hm5C, adenosine dimethyltransfer (m6Am) [9] as well as uridylation, pseudouridylation, removal of the noncanonical NAD+ cap, the biosynthesis of 5-methylaminomethyl-2-thiouridine (mnm(5)s(2)U) of tRNAs, and many others (Figure 1). All their functions are embedded in the transition from RNAs to proteins and are most likely important for the regulation of RNA localization, structure, stability, processing, ribosome assembly, and translational events. Modifiers and readers are believed to play important roles especially for abiotic stress responses, plant acclimation, and developmental processes [24].
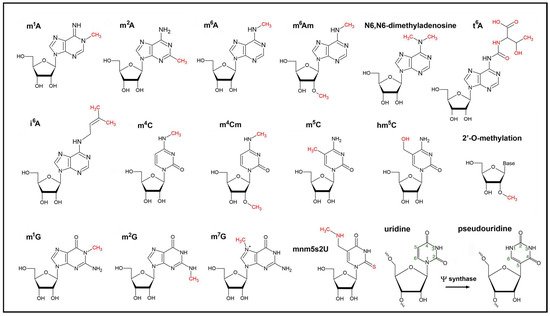
Figure 1. Chemical formulas of modified nucleotides found within chloroplast RNAs. The epitranscriptomic modifications are catalyzed by writers and are highlighted in red. The isomerization of uridine to pseudouridine (lower right) within RNAs is catalyzed by the Ψ synthase. The extra hydrogen bond at position N1 of pseudouridine stabilizes base pairing and RNA structure.
Our knowledge about the function of chloroplast epitranscriptomic activities is still in its infancy. Forthcoming state-of-the-art ‘omics’ technologies will considerably improve the monitoring of RNA modification networks at the genomic scale in the plant genetic compartments and will certainly shed light on the function and regulation of epitranscriptomic players.
2. Methods for the Detection of RNA Modification Marks
Immunoprecipitation of modified RNAs in combination with high-throughput sequencing, mass spectrometry-based techniques, and nanopore sequencing are the three most common approaches for the transcriptome-wide detection and quantification of epitranscriptomic marks. Some of the methods are further combined with chemical or enzymatic steps to identify signature-based marks in RNA molecules [25]. However, it is important to consider as mentioned below that each system has its intrinsic strengths and limitations.
2.1. Antibody-Based Approaches and Next-Generation Sequencing
Since antibodies are available for m1A, m6A, and m5C and other base modifications, several studies of the cellular epitranscriptome in Arabidopsis focused on immunological approaches, such as the predominant N6A-methylated RNA immunoprecipitation sequencing (MeRIP/m6A-seq). This technique requires significant quantities of RNAs, high quality antibodies with little cross reactivity, and involves immunoprecipitation of about ~100 nucleotides-long RNA fragments using m6A-specific antibodies, followed by sequencing of the immunoprecipitated fragments or the corresponding cDNAs [26]. However, this approach has several limitations with respect to the resolution and the specificity of the antibody. For example, antibodies for m6A can potentially also detect a second base modification, N6,2-O-dimethyladenosine (m6Am), which is located at the 5’ end of transcripts. In addition, the mandatory fragmentation of RNAs for library constructions may result in the underrepresentation of m6A sites [27]. To overcome these drawbacks, new approaches have been developed [28][29][30][31][32]. For example, m6A individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP) can be used to induce specific mutational signatures that allow for the precise identification of m6A residues in RNA molecules. In this approach, antibodies raised against m6A are UV-crosslinked to RNAs and subsequent reverse transcription of crosslinked RNAs results in a precise pattern of mutations or truncations in the cDNA. These signatures are computationally identified and allow mapping of m6A residues at single-nucleotide resolution [33].
Sequencing approaches in epitranscriptomics rely on cDNAs as template. A weak point of this method is that some of the epitranscriptomic information is lost when immunoprecipitated RNA is reverse transcribed. However, some of the modification marks, such as m1A, m3C, and m1G, can easily be detected because they induce reverse transcription errors or termination, as compared to their unmodified sites. For the detection of m5C, RNA can also be treated with sodium bisulfite prior reverse transcription. This chemical compound deaminates cytosine to uracil resulting in a thymine during reverse transcription. In contrast to the unmethylated cytosine, the m5C methylation is not prone to this deamination and thus can be detected.
2.2. Mass Spectrometric Approaches
The prerequisite for classical mass spectrometry in the detection of diverse RNA modifications including m6A is based on the complete enzymatic digestion of the RNA into individual nucleotides or nucleosides followed by various LC–MS/MS (liquid chromatography coupled to tandem mass spectrometry)-based methods [25]. After ionization the ions are detected based on their mass-to-charge ratio (m/z) to estimate their molecular mass and abundance. The modified RNA digestion products can readily be identified due to an increased molecular mass compared with the unmodified standards. This method plays an important role in the discovery, simultaneous detection, and quantification of many different RNA modifications. However, despite its high sensitivity, accuracy, and low detection limit in the fmol to amol range, co-eluting compounds with the identical masses, such as the modifications m1G and m2G, cannot be distinguished. Importantly, due to the complete digestion all information about the sequence context and co-occurrence of modifications is entirely lost. To overcome this shortcoming, RNA molecules are partially digested using specific RNases. The resulting RNA oligonucleotides are then analyzed by MS/MS and the covalent modifications are recognized with single-nucleotide resolution based on the molecular mass shift. A comparison with mass spectra provided by a sequence database enables the determination of the precise position and nature of modifications within the oligonucleotide that can be determined using a computational platform for high-throughput data mining [34].
2.3. Nanopore Sequencing
Nanopore direct RNA sequencing (DRS) can be applied to measure and quantify many but not all RNA methylations at defined positions without fragmentation or amplification. This technology relies on a protein nanopore that resides in a membrane through which an electrical current is created. The RNA sequence can be identified by the magnitude of signals transmitted when intact RNAs pass through the nanopore by a motor protein [35]. The use of nanopore sequencing was also successfully applied in revealing full-length mRNAs, mapping of the 5’ cap, the position, and estimated length of the poly(A) tail, different patterns of alternative splicing, and sites of internal RNA cleavage in Arabidopsis. Moreover, novel examples of intronic alternative polyadenylation that potentially modulates gene functions were identified using this technique.
3. The Cellular m6A RNA Epitranscriptome
3.1. m6A Methylation of Nuclear-Derived RNAs Related to Chloroplast Functions
Recent data revealed that m6A methylation marks in nuclear-derived RNAs are often related to chloroplast function, thus we will provide a brief overview of the nuclear/cytoplasmic m6A methylome with a focus on this organelle (Figure 2). With respect to RNA methylation, such as the most widespread adenosine methylation at the N6 position (m6A), the mainly nuclear localized modifiers (methyltransferases and demethylases) and mostly cytoplasmic interpreters are called writers, erasers, and readers, respectively [36]. The nuclear m6A writer complex consists of several conserved and essential components in eukaryotes, including the methyltransferases MTA and MTB, the splicing factor FKBP12 Interacting Protein37 (FIP37), VIRILIZER (VIR), and the conserved E3 ubiquitin ligase HAKAI [37]. DRS and miCLIP were recently applied to examine the epitranscriptome of the leaky vir-1 mutant defective in nuclear m6A methylation and VIR-complemented lines in Arabidopsis [32]. Frequent cleavage and polyadenylation of the mRNA encoding a chloroplast envelope-bound plant homeodomain transcription factor (PTM) with transmembrane domains was found. PTM (AT5G35210) resides in the outer chloroplast membrane and was suggested to be involved in retrograde signaling upon cleavage of a C-terminal transmembrane domain that sequesters it to the chloroplast [32][38]. Cleavage and polyadenylation of the ptm intron 10 terminates transcription prior to a sequence encoding the transmembrane domain, consequently bypassing established retrograde control [32]. 17,491 sites with restored m6A modifications in the VIR-complemented line were identified. The AAm6ACU and AAm6ACA motifs were confirmed to be the most frequently detected m6A marks in Arabidopsis [39][40][41]. Several sites associated with the motif AGm6AUU were also detected, raising the possibility that a C following m6A is not a constant feature of the Arabidopsis m6A code [32]. The preferential 3’ UTR localization of m6A in cytoplasmic mRNAs was also confirmed. The differential error sites were exclusively found in this region and no enrichment over stop codons was identified using both, miCLIP or DRS. Strikingly, the impact of m6A loss on pre-mRNA processing was determined and a clear defect in RNA 3’ end formation in vir-1 was observed [32]. 3579 genes with an altered 3’ position profile in the vir-1 mutant were identified. For instance, the prpl34 mRNA, which encodes a chloroplast ribosomal protein, is methylated in at least two positions in the 3’ UTR and displays an increase in alternative polyadenylation at a proximal poly(A) site in the vir-1 mutant. These findings suggest that changes in 3’ end poly(A) position of RNAs in the vir-1 mutant may result directly from the loss of m6A and implies a crucial role of the cellular m6A methylome in plastid gene expression.
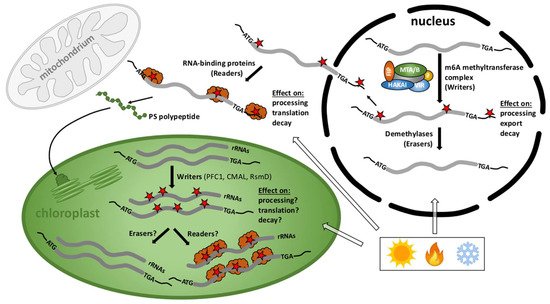
Figure 2. Cellular scenario of the RNA methylome in plants. m6A methylation and demethylation of nuclear-derived RNAs mainly takes place in the nucleus close to start and stop codons and in the 3’ UTRs and participate in the processing, stability, and localization of RNAs whereas m6A readers function mostly in the cytoplasm. m6A methylated transcripts predominantly encode chloroplast proteins important for gene expression, photosynthesis, and other plastid functions. Only three 16S rRNA writers–PFC1 for m6A, CMAL for m4Cm, and RsmD for m2G methylations-have been described in chloroplasts but writers for mRNAs, as well as erasers and readers are entirely unknown. The activity of these modifiers and interpreters is presumably crucial for the fate of transcripts important for plant development, stress responses, and acclimation processes upon environmental changes. Red stars: RNA methylation marks; orange clouds: readers.
Conflicting results in the Arabidopsis epitranscriptome, such as the enrichment of the m6A distribution within the plant mRNA molecules, can be solved using state-of-the-art approaches and new technologies. Still, it is important to restate that the effects of m6A are challenging and possibly condition-dependent. To date, many chloroplast-related, nucleus encoded transcripts that carry m6A were identified. Regardless of nucleus- or chloroplast-derived transcripts, a compelling connection between m6A RNA modifications and chloroplast and/or acclimation functions in plants seems to be clear. Despite the obvious progress summarized here, the exact understanding of how m6A modifications regulate the function of nucleus-encoded RNAs that encode chloroplast proteins remains a future challenge (Figure 2).
In general, the m6A modification is subjected to dynamic regulation in both development and response to cellular stimuli and ever-changing conditions in eukaryotes [25][42]. Although m6A appears to be the most abundant internal RNA modification of plants, the m6A pattern and its regulation in humans is by far much better investigated than in plants [37]. Importantly, a relatively high proportion of nuclear/cytoplasmic transcripts encoding photosynthesis-related proteins have been shown to undergo m6A modifications implying important roles in chloroplast functions. The m6A modifications generally play critical roles in many areas of the plant life [42][43]. Similar to the animal system the m6A:A ratio is 1.5% in young Arabidopsis seedlings [44]. Elucidation of the function of the m6A RNA modification is a challenging and growing field in plant RNA research [45]. Rapidly evolving methodological approaches will allow us to increase our understanding about the function and regulation of m6A in plants, which certainly will contribute to improve our knowledge about cellular functions, developmental cycles, and acclimation processes related to chloroplast functions.
The m6A methylome in plants was first identified in two accessions of A. thaliana, Can-0 and Hen-16, two wild-collected lines from areas that vary drastically in photosynthetically active radiation values. However, the m6A modifications were shown to be remarkably conserved across these two lines. Surprisingly, m6A was enriched not only within the 3’ UTRs and stop codons but also around the start codons, a feature only observed in plant RNAs [39]. The consensus recognition sequence of nuclear/cytoplasmic transcripts has been described as RRm6ACH in the epitranscriptome of mammals, where R = G > A and H = U > A > C [26][46]. Interestingly, in plant RNAs new conserved motifs were found, indicating the presence of distinct target sequence motifs for m6A target-methylations [21][39]. For example, the URUAY (R = G > A,Y = U > A) m6A methylation motif is plant specific and was shown to have a role in RNA stabilization [47]. In both, Can-0 and Hen-16, gene ontology unveiled many biological pathways related to chloroplast functions. In particular, more than 60% of cytoplasmic transcripts containing m6A in both, start and stop codons and about 40% of those carrying the modification only in the start site were highly associated with photosynthetic functions. A complete list can be accessed in a previous work [39].
Differential m6A patterns of cytoplasmic transcripts across different organs were also investigated in Arabidopsis [40]. More than 70% of the transcripts were m6A modified in leaves, flowers, and roots. The consensus sequence RRm6ACH was found in over 75% of the transcripts, but only one dominant peak of m6A enrichment was identified around the 3’ UTR and stop codons in the Arabidopsis transcriptome. Notably, all three organs analyzed share about 290 m6A-methylated transcripts and their coding proteins are mostly located in the chloroplast. Most interestingly, differential m6A methylation among leaves, flowers, and roots showed that green leaves had the highest extent of m6A methylation among the three organs. These transcripts are mainly related to photosynthesis, regulation of transcription, and stress response. Highly methylated transcripts presented in leaves and roots had specific functions related to the respective organs—photosynthesis in leaves and transport in roots [40].
The extent of m6A methylation was also compared to the levels of the respective transcripts in three organs of Arabidopsis. Most of the highly expressed transcripts were less modified by m6A when compared with transcripts expressed at low level. This observation implies an important function of m6A in regulation of RNA levels and/or stability in plant cells. Low level transcripts may require a relatively higher extent of m6A modifications to maintain RNA stability in the cells and vice versa [40]. A role of m6A in the stabilization of mRNAs under salt stress has been reported in Arabidopsis. In this case, m6A is dynamically added to salt-stress-related transcripts to protect RNAs from degradation [48]. The process of flowering, for instance, is delayed in alkbh10b mutant plants, which lack an m6A eraser. This phenotype can be explained by the destabilization of transcripts involved in flowering transition when m6A modifications are not reversed [49]. Therefore, the exact mechanisms of regulation involving m6A in plants are far from being identified. Why under certain conditions m6A stabilizes or destabilizes a specific transcript is still to be determined. So far, the dependence of the stability of nuclear-encoded RNAs associated with plastid functions also remains an open question. Whether and how m6A methylation in the nucleus/cytoplasmic system reshapes the proteome or modulates gene expression within the chloroplast and vice versa remains elusive.
3.2. General Aspects on m6A Methylation Marks in Chloroplast RNAs
Virtually nothing is known about m6A epitranscriptome players in chloroplasts. In contrast to nuclear/cytoplasmic systems, only one chloroplast m6A RNA writer has been described [50] but erasers and readers are yet to be discovered (Figure 2). The m6A methylome was studied in Arabidopsis chloroplasts and mitochondria [42][51]. mtRNAs in both Arabidopsis and cauliflower undergo N6-adenosine methylation modifications with an occurrence of about 4–5 m6A sites per 1000 adenosine residues. Several m6A modifications were detrimental for translation, while a single modification in the start codon suggested an enhancement in the translatability of the mitochondrial transcript [51].
Remarkably, chloroplast transcripts are highly m6A methylated, implying important roles in photosynthesis and/or plastid gene expression [42][43]. Over 98% of chloroplast transcripts were chemically modified by m6A, which is by far much more than the modification status found in the nuclear transcriptome (73%). Furthermore, about 4.6 to 5.8 m6A sites per transcript were found in the chloroplast but only about 1.4 to 2.0 sites per transcript in the cytoplasm, again emphasizing an important link between m6A modifications and chloroplast functions [41]. The most modified transcripts found in this analysis were associated with chloroplast rRNA and mRNAs.
The previously observed dominant m6A enrichment within the 3´ UTR and near stop codons in nuclear-derived mRNAs was not observed in the chloroplast. Instead, m6A peaks were found evenly distributed in chloroplast transcripts with higher methylations in exons when compared to introns, suggesting that the regulatory mechanisms may be different between the nucleus/cytoplasm and chloroplast systems. The translation and stability of chloroplast transcripts are commonly regulated by factors acting on 5’ and 3’ UTRs. In addition, degradation of plastid RNAs is thought to be initiated by endonucleolytic cleavages [4][52]. Thus, it is likely that m6A methylation in conjunction with RNA-binding proteins controls the fate of mRNAs in the plant organelle at multiple levels.
Most surprisingly, many m6A consensus sequences in the chloroplasts and mitochondria share homology to those in mammals and plant nuclear transcritptomes, indicating an evolutionary related process [41][51]. The two most common sequence motifs found in the Arabidopsis chloroplast transcriptome were GGm6ACC and GGm6ACU. The methylation extent compared to RNA levels in the chloroplast was similar to that found in the nuclear-derived RNAs in Arabidopsis, as most of the highly expressed plastid transcripts were less modified by m6A, and vice versa, corroborating a related or analogous development. However, tissue-specific deviations were also observed. For example, in root amyloplasts the moderately expressed transcripts were more methylated and those expressed at lower or higher levels carried less m6A modifications [41].
4. tRNA Modifications
tRNAs are vital components of the translation machinery and more than 100 tRNA modifications are known so far, which all seem to be associated with translation efficiency, accuracy, and preventing ribosomal frameshifting. Modifications are often conserved and occur at specific sites of distinct chloroplast tRNAs, such as m7G at tRNA(Met) [53], i6A and m1G at position 37 of tRNA(Cys) [54], t6A, m2G and m7G at tRNA(Ile) [55], m7G at tRNA(Leu) [56] and m2A at tRNA(Met) [57], pseudouridylation (see below), and many others. Furthermore, hypermodifications in anti-codon loops are often important for decoding the genetic code. A number of studies have shown that tRNA modifications influence various developmental processes and functions in the stress response in distinct organisms, but little attention has been drawn to the role of diverse tRNA modifications in chloroplasts [58].
4.1. 5-Methylaminomethyl-2-Thiouridine Modification
To read all possible 64 triple codons, a minimum of 32 tRNAs is needed. However, plant plastids contain less than 32 distinct tRNAs, suggesting that tRNAs with U in their wobble position might pair with any of the four bases at the third position of the codon via superwobble [59]. The base pairing selectivity at the wobble position, that is to say the stringency or flexibility of the anticodon of tRNAs, is regulated by posttranscriptional modifications of the wobble U (U34) and has been shown to be crucial for correct and efficient translation [60][61][62] as well as for preventing ribosomal frameshifting [63]. One of these wobble nucleoside modifications is the 5-methylaminomethyl-2-thiouridine or mnm5s2U.
The bacterial glucose-inhibited division (gid) operon encodes the two GidA and GidB enzymes essential for the biosynthesis of mnm5s2U of tRNAs and the S-adenosyl-L-methionine (SAM)-dependent methylation of the 16S rRNA in the highly conserved 530 loop important for ribosomal function, respectively [64]. Mutant analysis has shown that GidA and GidB activities are important for stress response, growth, morphology, antibiotic resistance, and bacterial pathogenesis [64].
In E. coli for example, the mnm5s2U modification is a two-step process, in which U34 of the tRNA is first modified to 5-carboxymethylaminomethyl-2-thiouridine (cmnm5s2U) by TrmE (also known as tRNA modification E MnmE) together with GidA [65][66] and then decarboxylated to nm5s2U and methylated in a S-adenosyl-L-methionine- (SAM)-dependent manner to produce mnm5s2U by the enzyme TrmC (also called MnmC) [67]. GidA and the GTP-binding protein MnmE, form a heterotetrameric α2β2 complex consisting of two homodimers in order to bind and to interdependently modify particular tRNAs at the wobble uridine base U34 of the first anticodon position in FAD-and GTP-dependent reactions to form mnm5s2U [68].
Near-isogenic lines generated by introgression and mutants in rice exhibited a pleiotropic phenotype with reduced chloroplast protein levels and altered gene expression that highly affects retrograde signaling [69]. Map-based cloning revealed that the allele PLEIOTROPIC DEVELOPMENTAL DEFECTS (PDD) is responsible for the phenotype and closely related to plant and cyanobacterial TrmE proteins. PDD is preferentially expressed in photosynthetic tissue. As in bacteria, the rice homolog is able to form dimers in vivo and to exhibit GTPase activity [69]. The natural variation form of PDD, called PDDOL, containing a 6-bp deletion as well as 28 SNPs in its ORF, was incapable of forming homodimers in introgression lines and showed a reduced GTPase activity. NIL-PDDOL plants showed multiple developmental defects accompanied by decreased levels of proteins involved in photosynthesis and ribosome biogenesis. Investigations of the tRNA modifications by LC-MS/MS revealed that modification levels of mnm5s2U were significantly reduced in NIL-PDDOL as compared to the WT.
Moreover, altered chloroplast transcription and translation in PDDOL seems to activate retrograde signaling, as expression levels of the photosynthesis-associated nuclear genes were significantly reduced in PDDOL. While the function and biosynthesis of the mnm5s2U modification in bacteria seems to be relatively well understood, the roles of mnm5s2U in chloroplasts remains elusive.
It is very interesting that natural variations of a chloroplast tRNA-modifying enzyme led to severe pleiotropic defects that not only function in chloroplast biogenesis but also in plant development and demonstrates a fast-evolving plastid RNA metabolism despite its conserved mechanism [4]. Whether the plant TrmE also forms a heterotetramer in association with a GidA homolog remains to be shown. Remarkably, the Arabidopsis genome encodes a highly conserved GidA homolog (AT2G13440) which is considered to be located in chloroplasts when using several prediction servers (aramemnon.uni-koeln.de, accessed on 23.07.2021). However, a functional characterization and experimental investigations of the capability of the GidA homolog to form functional tetrameric complexes with the plant TrmE are lacking. Addressing these points in future studies will certainly contribute to improve our understanding of mnm5s2U in chloroplasts.
4.2. i6A Modification
The 37th base of a subset of tRNAs next to the anticodon can be modified by bulky additions including N6-isopentenyl adenosine (i6A) in all kingdoms of life. The i6A modification can be modified further to 2-methyl-thio-N6-isopentenyladenosine (ms2i6A) and is believed to stabilize the Watson-Crick base pairing by base stacking. The isopentenylation is catalyzed by a conserved isopentenyl transferase important for translation accuracy, efficiency, and non-sense suppression [70].
The isopentenylation is also conserved at position 37 of chloroplast cysteine tRNAs carrying a GCA anticodon. It has been shown that the chloroplast tRNACys isoacceptor at position 37 stimulates read-through over UGA stop codons and thus has been considered as a natural UGA stop codon suppressor [54].
References
- Stern, D.B.; Goldschmidt-Clermont, M.; Hanson, M.R. Chloroplast RNA metabolism. Annu. Rev. Plant Biol. 2010, 61, 125–155.
- Stoppel, R.; Lezhneva, L.; Schwenkert, S.; Torabi, S.; Felder, S.; Meierhoff, K.; Westhoff, P.; Meurer, J. Recruitment of a ribosomal release factor for light-and stress-dependent regulation of petB transcript stability in Arabidopsis chloroplasts. Plant Cell 2011, 23, 2680–2695.
- Stoppel, R.; Meurer, J. Complex RNA metabolism in the chloroplast: An update on the psbB operon. Planta 2013, 237, 441–449.
- Manavski, N.; Schmid, L.M.; Meurer, J. RNA-stabilization factors in chloroplasts of vascular plants. Essays Biochem. 2018, 62, 51–64.
- Zou, M.; Mu, Y.; Chai, X.; Ouyang, M.; Yu, L.J.; Zhang, L.; Meurer, J.; Chi, W. The critical function of the plastid rRNA methyltransferase, CMAL, in ribosome biogenesis and plant development. Nucleic Acids Res. 2020, 48, 3195–3210.
- Stoppel, R.; Meurer, J. The cutting crew-ribonucleases are key players in the control of plastid gene expression. J. Exp. Bot. 2012, 63, 1663–1673.
- Lange, H.; Sement, F.M.; Canaday, J.; Gagliardi, D. Polyadenylation-assisted RNA degradation processes in plants. Trends Plant Sci. 2009, 14, 497–504.
- Schuster, G.; Stern, D. Chapter 10 RNA Polyadenylation and Decay in Mitochondria and Chloroplasts. Prog. Mol. Biol. Transl. Sci. 2009, 85, 393–422.
- Manduzio, S.; Kang, H. RNA methylation in chloroplasts or mitochondria in plants. RNA Biol. 2021, in press.
- Wang, X.; An, Y.; Xu, P.; Xiao, J. Functioning of PPR Proteins in Organelle RNA Metabolism and Chloroplast Biogenesis. Front. Plant Sci. 2021, 12, 1.
- Cho, W.K.; Geimer, S.; Meurer, J. Cluster analysis and comparison of various chloroplast transcriptomes and genes in Arabidopsis thaliana. DNA Res. 2009, 16, 31–44.
- Lee, K.; Kang, H. Roles of organellar RNA-binding proteins in plant growth, development, and abiotic stress responses. Int. J. Mol. Sci. 2020, 21, 4548.
- Manavski, N.; Torabi, S.; Lezhneva, L.; Arif, M.A.; Frank, W.; Meurer, J. HIGH CHLOROPHYLL FLUORESCENCE145 Binds to and Stabilizes the psaA 5′ UTR via a Newly Defined Repeat Motif in Embryophyta. Plant Cell. 2015, 27, 2600–2615.
- Anreiter, I.; Mir, Q.; Simpson, J.T.; Janga, S.C.; Soller, M. New Twists in Detecting mRNA Modification Dynamics. Trends Biotechnol. 2021, 39, 72–89.
- Kadumuri, R.V.; Janga, S.C. Epitranscriptomic Code and Its Alterations in Human Disease. Trends Mol. Med. 2018, 24, 886–903.
- Shen, L.; Liang, Z.; Wong, C.E.; Yu, H. Messenger RNA Modifications in Plants. Trends Plant Sci. 2019, 24, 328–341.
- Wiener, D.; Schwartz, S. The epitranscriptome beyond m6A. Nat. Rev. Genet. 2020, 22, 119–131.
- Burgess, A.; David, R.; Searle, I.R. Deciphering the epitranscriptome: A green perspective. J. Integr. Plant Biol. 2016, 58, 822–835.
- Vandivier, L.E.; Gregory, B.D. New insights into the plant epitranscriptome. J. Exp. Bot. 2018, 69, 4659–4665.
- Liang, Z.; Riaz, A.; Chachar, S.; Ding, Y.; Du, H.; Gu, X. Epigenetic Modifications of mRNA and DNA in Plants. Mol. Plant 2020, 13, 14–30.
- Parker, M.T.; Knop, K.; Simpson, G.G. Making a mark: The role of RNA modifications in plant biology. Biochem. (Lond). 2020, 42, 26–30.
- Kleine, T.; Nägele, T.; Neuhaus, H.E.; Schmitz-Linneweber, C.; Fernie, A.R.; Geigenberger, P.; Grimm, B.; Kaufmann, K.; Klipp, E.; Meurer, J.; et al. Acclimation in plants – the Green Hub consortium. Plant J. 2020, 106, 23–40.
- Sandoval, R.; Boyd, R.D.; Kiszter, A.N.; Mirzakhanyan, Y.; Santibańez, P.; Gershon, P.D.; Hayes, M.L. Stable native RIP9 complexes associate with C-to-U RNA editing activity, PPRs, RIPs, OZ1, ORRM1 and ISE2. Plant J. 2019, 99, 1116–1126.
- Hu, J.; Manduzio, S.; Kang, H. Epitranscriptomic RNA methylation in plant development and abiotic stress responses. Front. Plant Sci. 2019, 10, 500.
- Mongan, N.P.; Emes, R.D.; Archer, N. Detection and analysis of RNA methylation. F1000Research 2019, 8, 559.
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646.
- McIntyre, A.B.R.; Gokhale, N.S.; Cerchietti, L.; Jaffrey, S.R.; Horner, S.M.; Mason, C.E. Limits in the detection of m6A changes using MeRIP/ m6A-seq. Sci. Rep. 2020, 10, 1–15.
- Garcia-Campos, M.A.; Edelheit, S.; Toth, U.; Safra, M.; Shachar, R.; Viukov, S.; Winkler, R.; Nir, R.; Lasman, L.; Brandis, A.; et al. Deciphering the “m6A Code” via Antibody-Independent Quantitative Profiling. Cell 2019, 178, 731–747.e16.
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772.
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun. 2019, 10, 1–9.
- Meyer, K.D. DART-seq: An antibody-free method for global m6A detection. Nat. Methods 2019, 16, 1275–1280.
- Parker, M.T.; Knop, K.; Sherwood, A.V.; Schurch, N.J.; Mackinnon, K.; Gould, P.D.; Hall, A.J.W.; Barton, G.J.; Simpson, G.G. Nanopore direct RNA sequencing maps the complexity of arabidopsis mRNA processing and m6A modification. Elife 2020, 9.
- Grozhik, A.V.; Linder, B.; Olarerin-George, A.O.; Jaffrey, S.R. Mapping m6A at individual-nucleotide resolution using crosslinking and immunoprecipitation (MiCLIP). In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1562, pp. 55–78.
- Sarkar, A.; Gasperi, W.; Begley, U.; Nevins, S.; Huber, S.M.; Dedon, P.C.; Begley, T.J. Detecting the epitranscriptome. Wiley Interdiscip. Rev. RNA 2021, e1663, in press.
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RN A sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206.
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624.
- Arribas-Hernández, L.; Brodersen, P. Occurrence and functions of m6A and other covalent modifications in plant mRNA. Plant Physiol. 2020, 182, 79–96.
- Feng, P.; Guo, H.; Chi, W.; Chai, X.; Sun, X.; Xu, X.; Ma, J.; Rochaix, J.D.; Leister, D.; Wang, H.; et al. Chloroplast retrograde signal regulates flowering. Proc. Natl. Acad. Sci. USA 2016, 113, 10708–10713.
- Luo, G.Z.; Macqueen, A.; Zheng, G.; Duan, H.; Dore, L.C.; Lu, Z.; Liu, J.; Chen, K.; Jia, G.; Bergelson, J.; et al. Unique features of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 2014, 5, 1–8.
- Wan, Y.; Tang, K.; Zhang, D.; Xie, S.; Zhu, X.; Wang, Z.; Lang, Z. Transcriptome-wide high-throughput deep m6A -seq reveals unique differential m6A methylation patterns between three organs in Arabidopsis thaliana. Genome Biol. 2015, 16, 1–26.
- Wang, Z.; Tang, K.; Zhang, D.; Wan, Y.; Wen, Y.; Lu, Q.; Wang, L. High-throughput m6A -seq reveals RNA m6A methylation patterns in the chloroplast and mitochondria transcriptomes of Arabidopsis thaliana. PLoS ONE 2017, 12, e0185612.
- Reichel, M.; Köster, T.; Staiger, D. Marking RNA: m6A writers, readers, and functions in Arabidopsis. J. Mol. Cell Biol. 2019, 11, 899–910.
- Yue, H.; Nie, X.; Yan, Z.; Weining, S. N6-methyladenosine regulatory machinery in plants: Composition, function and evolution. Plant Biotechnol. J. 2019, 17, 1194–1208.
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008, 20, 1278–1288.
- Lockhart, J. The story continues: Following the fate of m6A marks in the eukaryotic transcriptome. Plant Cell 2018, 30, 1385–1386.
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206.
- Wei, L.H.; Song, P.; Wang, Y.; Lu, Z.; Tang, Q.; Yu, Q.; Xiao, Y.; Zhang, X.; Duan, H.C.; Jia, G. The m6A reader ECT2 controls trichome morphology by affecting mRNA stability in Arabidopsis. Plant Cell 2018, 30, 968–985.
- Anderson, S.J.; Kramer, M.C.; Gosai, S.J.; Yu, X.; Vandivier, L.E.; Nelson, A.D.L.; Anderson, Z.D.; Beilstein, M.A.; Fray, R.G.; Lyons, E.; et al. N6-Methyladenosine Inhibits Local Ribonucleolytic Cleavage to Stabilize mRNAs in Arabidopsis. Cell Rep. 2018, 25, 1146–1157.e3.
- Duan, H.C.; Wei, L.H.; Zhang, C.; Wang, Y.; Chen, L.; Lu, Z.; Chen, P.R.; He, C.; Jia, G. ALKBH10B is an RNA N6-methyladenosine demethylase affecting Arabidopsis floral transition. Plant Cell 2017, 29, 2995–3011.
- Tokuhisa, J.G.; Vijayan, P.; Feldmann, K.A.; Browse, J.A. Chloroplast development at low temperatures requires a homolog of DIM1, a yeast gene encoding the 18S rRNA dimethylase. Plant Cell 1998.
- Murik, O.; Chandran, S.A.; Nevo-Dinur, K.; Sultan, L.D.; Best, C.; Stein, Y.; Hazan, C.; Ostersetzer-Biran, O. Topologies of N6-adenosine methylation (m6A) in land plant mitochondria and their putative effects on organellar gene expression. Plant J. 2020, 101, 1269–1286.
- Monde, R.A.; Schuster, G.; Stern, D.B. Processing and degradation of chloroplast mRNA. Biochimie 2000, 82, 573–582.
- Canaday, J.; Guillemaut, P.; Weil, J.H. The nucleotide sequences of the initiator transfer RNAs from bean cytoplasm and chloroplasts. Nucleic Acids Res. 1980, 8, 999–1008.
- Urban, C.; Beier, H. Cysteine tRNAs of plant origin as novel UGA suppressors. Nucleic Acids Res. 1995, 23, 4591–4597.
- Guillemaut, P.; Weil, J.H. The nucleotide sequence of the maize and spinach chloroplast isoleucine transfer RNA encoded in the 16S to 23S rDNA spacer. Nucleic Acids Res. 1982, 10, 1653–1659.
- Jakab, G.; Kis, M.; Pálfi, Z.; Solymosy, F. Nucleotide sequence of chloroplast tRNALeu/UA m7G/ from Chlamydomonas reinhardtii. Nucleic Acids Res. 1990, 18, 7444.
- McCoy, J.M.; Keene, N.M.; Jones, D.S. The nucleotide sequence of Scenedesmus obliquus chloroplast elongator methionine-accepting tRNA. Biochem. J. 1986, 238, 297–300.
- de Crécy-Lagard, V.; Jaroch, M. Functions of Bacterial tRNA Modifications: From Ubiquity to Diversity. Trends Microbiol. 2021, 29, 41–53.
- Alkatib, S.; Scharff, L.B.; Rogalski, M.; Fleischmann, T.T.; Matthes, A.; Seeger, S.; Schöttler, M.A.; Ruf, S.; Bock, R. The Contributions of Wobbling and Superwobbling to the Reading of the Genetic Code. PLoS Genet. 2012, 8, e1003076.
- Yokoyama, S.; Watanabe, T.; Muraot, K.; Ishikurat, H.; Yamaizumit, Z.; Nishimurat, S.; Miyazawa, T. Molecular mechanism of codon recognition by tRNA species with modified uridine in the first position of the anticodon (post-transcriptional modification/base pair/conformation/NMR). Proc. Natl. Acad. Sci. USA 1985, 82, 4905–4909.
- Krüger, M.K.; Pedersen, S.; Hagervall, T.G.; Sørensen, M.A. The modification of the wobble base of tRNA(Glu) modulates the translation rate of glutamic acid codons in vivo. J. Mol. Biol. 1998, 284, 621–631.
- Johansson, M.J.O.; Esberg, A.; Huang, B.; Björk, G.R.; Byström, A.S. Eukaryotic Wobble Uridine Modifications Promote a Functionally Redundant Decoding System. Mol. Cell. Biol. 2008, 28, 3301–3312.
- Brégeon, D.; Colot, V.; Radman, M.; Taddei, F. Translational misreading: A tRNA modification counteracts a +2 ribosomal frameshift. Genes Dev. 2001, 15, 2295–2306.
- Shippy, D.C.; Fadl, A.A. RNA modification enzymes encoded by the gid operon: Implications in biology and virulence of bacteria. Microb. Pathog. 2015, 89, 100–107.
- Cabedo, H.; Macián, F.; Villarroya, M.; Escudero, J.C.; Martínez-Vicente, M.; Knecht, E.; Armengod, M.E. The Escherichia coli trmE (mnmE) gene, involved in tRNA modification, codes for an evolutionarily conserved GTPase with unusual biochemical properties. EMBO J. 1999, 18, 7063–7076.
- Shi, R.; Villarroya, M.; Ruiz-Partida, R.; Li, Y.; Proteau, A.; Prado, S.; Moukadiri, I.; Benítez-Páez, A.; Lomas, R.; Wagner, J.; et al. Structure-function analysis of Escherichia coli MnmG (GidA), a highly conserved tRNA-modifying enzyme. J. Bacteriol. 2009, 191, 7614–7619.
- Hagervall, T.G.; Edmonds, C.G.; McCloskey, J.A.; Björk, G.R. Transfer RNA(5-methylaminomethyl-2-thiouridine)-methyltransferase from Escherichia coli K-12 has two enzymatic activities. J. Biol. Chem. 1987, 262, 8488–8495.
- Böhme, S.; Meyer, S.; Krüger, A.; Steinhoff, H.J.; Wittinghofer, A.; Klare, J.P. Stabilization of G domain conformations in the tRNA-modifying MnmE-GidA complex observed with double electron electron resonance spectroscopy. J. Biol. Chem. 2010, 285, 16991–17000.
- Liu, H.; Ren, D.; Jiang, L.; Li, X.; Yao, Y.; Mi, L.; Chen, W.; Mo, A.; Jiang, N.; Yang, J.; et al. A natural variation in pleiotropic developmental defects uncovers a crucial role for chloroplast tRNA modification in translation and plant development. Plant Cell 2020, 32, 2345–2366.
- Schweizer, U.; Bohleber, S.; Fradejas-Villar, N. The modified base isopentenyladenosine and its derivatives in tRNA. RNA Biol. 2017, 14, 1197–1208.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
03 Aug 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No