 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simona Bungău | + 1831 word(s) | 1831 | 2021-07-30 04:36:53 | | | |
| 2 | Catherine Yang | Meta information modification | 1831 | 2021-08-02 10:33:41 | | |
Video Upload Options
The astrocytes and neuronal cells are considered to be neuroprotective, whereas the infiltrating macrophages and activated microglia are considered to be neurotoxic. Therefore, multiple products of KP can have neuroprotective, neurotoxic, and immunomodulatory effects. QUIN, an excitotoxin, is reported to be the most significant among them, which results in the death of neurons.
1. Introduction
The greatest obstacle associated with neurodegenerative disorders is that they are incurable, and the deterioration is progressive with time and age. These pathologies vary in symptoms, pathological features, and drug candidates. Parkinson’s disease (PD), as well as Alzheimer’s disease (AD), are considered to be the most prevalent among the neurodegeneration-related disorders [1]. Recently, PD was reported to affect about 6.1 million people, in comparison to 2.5 million people in 1990 [2]. Accounting for the 21.7% of age standardized disease prevalence rate, the disease progressively accelerated during all these years. In 2016, PD led to 211,296 deaths around the globe [3]. The symptoms in PD include tremors, rigidity, bradykinesia, and disrupted posture, parallel to retardation in mental processing, speech problems, memory losses, and leaning inability.
The prime pathological features of the disease include loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and alleviated dopamine (DA) levels in the striatum [4][5]; however, the specific cause of the disease is still to be investigated. When the loss of dopaminergic neurons reaches 70% to 80%, it is marked with the appearance of associated signs and symptoms [2]. Therefore, this period (from the beginning of the loss of dopaminergic neurons in the brain to the identification of clinical signs and symptoms) marks the basis for development of effective treatment therapies for the disease. Despite the availability of numerous treatment options, no therapy is completely effective for this disorder. The therapies available for the disease are associated with hindrances, such as inability to cross the blood–brain barrier (BBB), side effects, limited life span, etc., alongside primary challenges such as the development of sensitive and reliable of biomarkers for disease detection [6][7]. Thus, there is a dire need to develop novel treatment options for PD, with greater specificity and selectivity for the disease targets and limited side effects.
The enzymes associated with KP have been recognized as reliable and promising target candidates for treatment of neurodegenerative diseases. Ageing and inflammation can alter the KP metabolism and availability of TRP [8], thus elevating the susceptibility towards age-dependent neurological disorders, including PD [9][10]. Chronic inflammation of the intestine, gut microbiota changes, and α-synuclein aggregation are the pivotal features of PD pathogenesis [11].
2. The Kynurenine Pathway
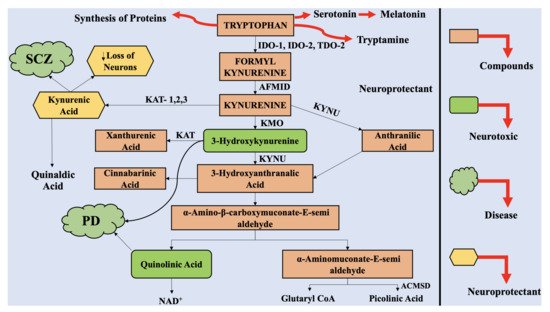
3. The Interaction of Kynurenine Pathway with the Central Nervous System
The physiological concentration of QUIN in the cerebrospinal fluid (CSF) and brain is approximately 100 nM, and it exhibits hormesis action. QUIN treatment results in elevation of NAD+ production, within the human neuronal cells, alongside enhanced proliferation of stem cells [34]. However, the concentration of QUIN is increased to 1200 nM in disease conditions, which can lead to acute, chronic, or progressively severe functions of neurons or death of neuronal cells by a minimum of nine varying processes [35]. At pathological concentrations, between 150 to 1200 nM, QUIN functions as NMDA receptor agonists. Previously, two studies reported the selective actions of QUIN on NMDA receptor, along with differences in actions, mediated by QUIN on NMDA, at particular neuronal areas [36]. Vandresen-Filho et al. confirmed the above statement and reported that the hippocampus, cerebral cortex, cerebellum, and striatum have varying susceptibility towards QUIN induced oxidative stress [37]. The neuronal cells contained in the striatum, hippocampus, and neocortex, exhibit sensitivity towards QUIN; however, the neurons present in the spinal cord and cerebellar region are comparatively less sensitive. These differences are associated with alterations in the configurations of the NMDA receptor [38].
It was depicted that the motor neuron could be completely protected from QUIN-mediated excitotoxicity by using NMDA receptor (NMDAR) antagonist combinations [39]. QUIN has the ability to cause elevated micro-environment glutamate levels and neurotoxicity by enhancing the release of glutamate by neuronal cells, blocking its astrocyte-mediated uptake and inhibiting astroglial glutamine synthetase enzyme [40].
Oxidative stress is induced in the astrocytes and neuronal cells by QUIN, which results in the death of the glial cells and neurons, by energy restriction. The phosphorylation of cellular structural proteins, such as glial fibrillary acidic protein (GFAP) in astrocytes and neurofilament in neuronal cells are elevated by QUIN, resulting in the destabilization of the cytoskeleton [41][42]. It has been reported that the tau phosphorylation in neuronal cells in humans is elevated by QUIN, which further co-localizes with hyperphosphorylated tau in neuronal cells in cortex of AD-affected neuronal tissue [42].
The cellular agents, which provide support to the neuronal cells, i.e., astrocytes, are also altered by QUIN, which also dysregulated astroglial functions and promote death of the glial cells [43]. As a result, dysregulated function and death of neuronal cells take place [28]. Furthermore, QUIN exerts proinflammatory effect on astrocytes, followed by generation of proinflammatory cytokines as well as chemokines, such as monocyte chemotactic protein 1 (MCP-1), IL-1β, and IL-8 in astrocytes [40]. The activity of glutamine synthetase is retarded by QUIN in a dose dependent manner in the astrocytes in humans, which leads to disruption of glutamine/glutamate cycle [40].
References
- Berg, D.; Postuma, R.B.; Bloem, B.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.M.; Hardy, J.; Lang, A.E. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov. Disord. 2014, 29, 454–462.
- Pingale, T.; Gupta, G.L. Current and emerging therapeutic targets for Parkinson’s disease. Metab. Brain Dis. 2021, 36, 13–27.
- Tepper, S.; Ashina, M.; Reuter, U.; Brandes, J.L.; Doležil, D.; Silberstein, S.; Winner, P.; Leonardi, D.; Mikol, D.; Lenz, R. Safety and efficacy of erenumab for preventive treatment of chronic migraine: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017, 16, 425–434.
- Angot, E.; Brundin, P. Dissecting the potential molecular mechanisms underlying α-synuclein cell-to-cell transfer in Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, S143–S147.
- Bartel, W.P.; Van Laar, V.S.; Burton, E.A. Chapter 23—Parkinson’s disease. In Behavioral and Neural Genetics of Zebrafish; Gerlai, R.T., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 377–412.
- George, J.; Mok, S.; Moses, D.; Wilkins, S.; Bush, A.I.; Cherny, R.A.; Finkelstein, D.I. Targeting the progression of Parkinson’s disease. Curr. Neuropharmacol. 2009, 7, 9–36.
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808.
- Erhardt, S.; Schwieler, L.; Imbeault, S.; Engberg, G. The kynurenine pathway in schizophrenia and bipolar disorder. Neuropharmacology 2017, 112, 297–306.
- Stetler, R.A.; Leak, R.K.; Gan, Y.; Li, P.; Zhang, F.; Hu, X.; Jing, Z.; Chen, J.; Zigmond, M.J.; Gao, Y. Preconditioning provides neuroprotection in models of CNS disease: Paradigms and clinical significance. Prog. Neurobiol. 2014, 114, 58–83.
- Török, N.; Török, R.; Szolnoki, Z.; Somogyvári, F.; Klivényi, P.; Vécsei, L. The genetic link between Parkinson’s disease and the kynurenine pathway is still missing. Parkinsons Dis. 2015, 2015, 474135.
- Houser, M.; Tansey, M. The gut-brain axis: Is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinsons Dis. 2017, 3, 3.
- Massudi, H.; Grant, R.; Guillemin, G.J.; Braidy, N. NAD+ metabolism and oxidative stress: The golden nucleotide on a crown of thorns. Redox Rep. 2012, 17, 28–46.
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine pathway, NAD+ synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 2020, 132, 110841.
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, aaf9794.
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100.
- Munn, D.H.; Mellor, A.L. IDO and tolerance to tumors. Trends Mol. Med. 2004, 10, 15–18.
- Dürr, S.; Kindler, V. Implication of indolamine 2,3 dioxygenase in the tolerance toward fetuses, tumors, and allografts. J. Leukoc. Biol. 2013, 93, 681–687.
- Mazarei, G.; Leavitt, B.R. Indoleamine 2,3 dioxygenase as a potential therapeutic target in Huntington’s disease. J. Huntingt. Dis. 2015, 4, 109–118.
- Widner, B.; Leblhuber, F.; Fuchs, D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J. Neural Transm. 2002, 109, 181–189.
- Mellor, A.L.; Baban, B.; Chandler, P.; Marshall, B.; Jhaver, K.; Hansen, A.; Koni, P.A.; Iwashima, M.; Munn, D.H. Cutting edge: Induced indoleamine 2, 3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J. Immunol. 2003, 171, 1652–1655.
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190.
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112.
- Jones, S.P.; Franco, N.F.; Varney, B.; Sundaram, G.; Brown, D.A.; De Bie, J.; Lim, C.K.; Guillemin, G.J.; Brew, B.J. Expression of the kynurenine pathway in human peripheral blood mononuclear cells: Implications for inflammatory and neurodegenerative disease. PLoS ONE 2015, 10, e0131389.
- Guillemin, G.J.; Cullen, K.M.; Lim, C.K.; Smythe, G.A.; Garner, B.; Kapoor, V.; Takikawa, O.; Brew, B.J. Characterization of the kynurenine pathway in human neurons. J. Neurosci. 2007, 27, 12884–12892.
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82.
- Guillemin, G.J.; Kerr, S.J.; Brew, B.J. Involvement of quinolinic acid in AIDS dementia complex. Neurotox. Res. 2005, 7, 103–123.
- Pierozan, P.; Biasibetti, H.; Schmitz, F.; Ávila, H.; Parisi, M.M.; Barbe-Tuana, F.; Wyse, A.T.S.; Pessoa-Pureur, R. Quinolinic acid neurotoxicity: Differential roles of astrocytes and microglia via FGF-2-mediated signaling in redox-linked cytoskeletal changes. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 3001–3014.
- Lim, C.K.; Fernandez-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2017, 155, 76–95.
- Chiarugi, A.; Meli, E.; Moroni, F. Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid. J. Neurochem. 2001, 77, 1310–1318.
- Ramírez-Ortega, D.; Ramiro-Salazar, A.; González-Esquivel, D.; Ríos, C.; Pineda, B.; Pérez de la Cruz, V. 3-Hydroxykynurenine and 3-hydroxyanthranilic acid enhance the toxicity induced by copper in rat astrocyte culture. Oxidative Med. Cell. Longev. 2017, 2017, 2371895.
- Goldstein, L.E.; Leopold, M.C.; Huang, X.; Atwood, C.S.; Saunders, A.J.; Hartshorn, M.; Lim, J.T.; Faget, K.Y.; Muffat, J.A.; Scarpa, R.C. 3-Hydroxykynurenine and 3-hydroxyanthranilic acid generate hydrogen peroxide and promote α-crystallin cross-linking by metal ion reduction. Biochemistry 2000, 39, 7266–7275.
- Grant, R.; Coggan, S.; Smythe, G. The physiological action of picolinic acid in the human brain. Int. J. Tryptophan Res. 2009, 2, IJTR-S2469.
- La Cruz, V.P.-D.; Carrillo-Mora, P.; Santamaría, A. Quinolinic acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int. J. Tryptophan Res. 2012, 5, IJTR-S8158.
- Jones, S.P.; Guillemin, G.J.; Brew, B.J. The kynurenine pathway in stem cell biology. Int. J. Tryptophan Res. 2013, 6, 57–66.
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxidative Med. Cell. Longev. 2013, 2013, 104024.
- Perkins, M.N.; Stone, T.W. Pharmacology and regional variations of quinolinic acid-evoked excitations in the rat central nervous system. J. Pharm. Exp. 1983, 226, 551–557.
- Vandresen-Filho, S.; Martins, W.C.; Bertoldo, D.B.; Mancini, G.; De Bem, A.F.; Tasca, C.I. Cerebral cortex, hippocampus, striatum and cerebellum show differential susceptibility to quinolinic acid-induced oxidative stress. Neurol. Sci. 2015, 36, 1449–1456.
- Kumar, U. Characterization of striatal cultures with the effect of QUIN and NMDA. Neurosci. Res. 2004, 49, 29–38.
- Chen, Y.; Brew, B.J.; Guillemin, G.J. Characterization of the kynurenine pathway in NSC-34 cell line: Implications for amyotrophic lateral sclerosis. J. Neurochem. 2011, 118, 816–825.
- Ting, K.K.; Brew, B.J.; Guillemin, G.J. Effect of quinolinic acid on human astrocytes morphology and functions: Implications in Alzheimer’s disease. J. Neuroinflamm. 2009, 6, 36.
- Pierozan, P.; Zamoner, A.; Soska, A.K.; Silvestrin, R.B.; Loureiro, S.O.; Heimfarth, L.; Mello e Souza, T.; Wajner, M.; Pessoa-Pureur, R. Acute intrastriatal administration of quinolinic acid provokes hyperphosphorylation of cytoskeletal intermediate filament proteins in astrocytes and neurons of rats. Exp. Neurol. 2010, 224, 188–196.
- Rahman, A.; Ting, K.; Cullen, K.M.; Braidy, N.; Brew, B.J.; Guillemin, G.J. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS ONE 2009, 4, e6344.
- Lee, M.-C.; Ting, K.K.; Adams, S.; Brew, B.J.; Chung, R.; Guillemin, G.J. Characterisation of the Expression of NMDA Receptors in Human Astrocytes. PLoS ONE 2010, 5, e14123.

