+1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carmen Perez de Nanclares Fernandez | + 2021 word(s) | 2021 | 2021-07-27 13:23:36 | | | |
| 2 | Beatrix Zheng | Meta information modification | 2021 | 2021-07-29 04:18:11 | | | | |
| 3 | Conner Chen | Meta information modification | 2021 | 2021-09-22 04:20:11 | | |
Video Upload Options
Recent studies implicate astrocytes in Alzheimer’s disease (AD); however, their role in pathogenesis is poorly understood. Astrocytes have well-established functions in supportive functions such as extracellular ionic homeostasis, structural support, and neurovascular coupling. However, emerging research on astrocytic function in the healthy brain also indicates their role in regulating synaptic plasticity and neuronal excitability via the release of neuroactive substances named gliotransmitters. Here, we review how this “active” role of astrocytes at synapses could contribute to synaptic and neuronal network dysfunction and cognitive impairment in AD.
1. Introduction
AD is the most common neurodegenerative disorder with the contribution of multiple cell types throughout the central nervous system. Alois Alzheimer in 1907 first described the histopathological characteristics of the disease with the presence of senile plaques (formed by the extracellular aggregation of amyloid β (Aβ) protein) and neurofibrillary tangles (formed by the destructuring of the microtubules due to hyperphosphorylation of the tau protein).
Even though the disease was described more than a century ago, its underlying causes are still unknown today. One of the most accepted theories is the amyoidogenic hypothesis, where an accumulation and deposition of oligomeric or fibrillary Aβ peptides occur. The central role of the Aβ peptide in AD pathogenesis has been supported by the fact that familial forms of the disease are caused by its overproduction. Likewise, the contribution of microtubule-associated protein tau in AD pathology has also been proposed. In this hypothesis, the presence of abnormally high levels of tau protein in the hyperphosphorylated state promotes the production of toxic oligomeric tau and paired helical filaments, which further assemble into toxic neurofibrillary tangles.
The most predominant and striking sign in an AD patient is the progressive decline in cognition, which is primarily due to the loss of neurons and synapses in the hippocampal formation and related areas. As expected, research on AD has focused mainly on neuronal cells, which allowed great insights on the neurocentric molecular pathways involved in these processes, such as protein degradation systems, post-translational modifications, and interactions with cytoskeletons. However, in the last three decades, growing evidence suggests that glial cells, in particular astrocytes, have a contributing role in AD pathophysiology. Astrocytes in the human brain play key roles in numerous functions within the central nervous system such as structural support, ionic balance of the extracellular space, neurotransmitter clearance at synapses, and modulation of synaptic signaling. Thus, it is hardly surprising that astrocytes have been implicated in the pathology of several neurodegenerative diseases, including AD. Numerous excellent reviews have discussed the role of astrocytes in AD in the context of altered inflammatory processes, amyloid clearance, and neurovascular coupling. The present review focuses instead on the role of dysregulated bidirectional communication of neurons and astrocytes in AD pathology.
2. Reactive Astrocytes in Neurodegenerative Diseases
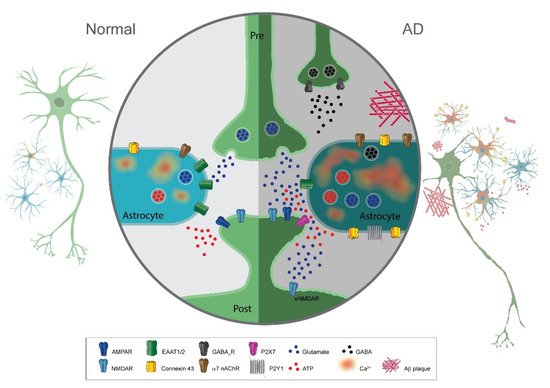
A hallmark sign of neurodegenerative disease is reactive gliosis with astrocytic morphological and functional changes. Astrocyte reactivity is initially characterized by the hypertrophy of soma and processes triggered by inflammatory molecules. In addition, there is an increased expression of intermediary filament proteins such as Glial Fibrillary Acidic Protein (GFAP) or vimentin. Many studies have shown that in AD, there is a prominent presence of reactive astrocytes, in particular, surrounding amyloid plaques. They are detected at the early phases of AD even before neuronal death and are found ubiquitously throughout disease progression. In addition to morphological changes, they display graded reactivity with heterogeneous gene expression, morphology, and function. Concerning this review, an important consequence of astrocyte reactivity in disease is the abnormal gliotransmitter release.
3. Altered Astrocyte Calcium Excitability in AD
There is great consensus among all available studies that in parallel with morphological changes and enhanced GFAP expression, spontaneous calcium signals in astrocytes are heightened in brains of AD animal models. This increase appears to be independent of neuronal activity, since the blockade of neuronal activity with the sodium channel blocker tetrodotoxin does not instigate changes in spontaneous calcium activity. This is in contrast to what is observed in normal aging, where no changes or even fewer spontaneous calcium oscillations occur.
Mechanistically, the increase in calcium activity in astrocytes may be mediated through the upregulation of neurotransmitter receptors in astrocytes such as the purinergic receptor P2Y1R, α7 nicotinic acetylcholine receptors (α7 nAChR), or metabotropic glutamate receptors mGluR5. Alternatively, Aβ plaques may bind and activate/inactivate α7 nAChR in astrocytes leading to hyperactivity in calcium signaling.
Since astrocytes can release gliotransmitters in response to intracellular calcium elevations, the upregulation of these neurotransmitter receptors in AD may trigger the dysregulated release of gliotransmitters such as glutamate to ultimately induce neurotoxicity.
4. Altered Glutamate Release and Uptake from Astrocytes in AD
Glutamate is the major excitatory transmitter in the central nervous system and has a large array of physiological functions including learning and memory.
Glutamate concentrations in the extracellular space are kept at low levels and tightly controlled by several mechanisms at the synapse. Perturbations to this regulatory system can lead to deleterious effects such as excess of extracellular glutamate, which can induce hyperexcitability in post-synaptic neurons to the point of excitotoxicity and cell death (cytotoxicity). It is usually accepted that the glutamate transporter activity of astrocytes is the primary mechanism responsible for clearing extracellular glutamate at synapses. The major type of glutamate transporter expressed in astrocytes is the sodium-dependent symporter GLT-1 (GLT-1), since it is responsible for around 90% of astrocytic glutamate uptake in the brain.
The bulk of evidence indicates a decrease in expression and activity of GLT-1 in AD, leading to cognitive deficits. Accordingly, the overexpression of GLT-1 in AD mice improved their cognitive functions and decreased pathology.
Astrocytes are not only involved in glutamate uptake via transporter activity, but they can also release glutamate as a gliotransmitter, most likely via a vesicular mechanism. Glutamate released by astrocytes binds to either presynaptic metabotropic glutamate receptors or extrasynaptically-located postsynaptic NMDA receptors (NR2B). The latter evokes the so-called slow inward currents (SICs). The depolarizing action of these SICs is hypothesized to modulate neural excitability to affect neuronal action potential firing.
Overall, several studies report an increase in astrocytic-mediated SICs in animal or experimental models of AD. The heightened release of glutamate from astrocytes may underlie the changes in synaptic function and plasticity observed in animal models of AD.
5. Altered ATP Release from Astrocytes
It is now well established that adenosine triphosphate (ATP) serves as an important neurotransmitter in the central and peripheral nervous system. ATP mediates fast and slow synaptic potentials via ligand-gated cationic channels (P2X receptors) and G protein-coupled receptors (P2Y receptors). It is also now apparent that ATP is released from astrocytes as a gliotransmitter.
Enhancement of ATP release has been shown in different models of AD. Several studies have proposed that connexin 43 (Cx43) in a hemichannel function mediates the Aβ peptide induction of ATP release from astrocytes. Thus, Cx43 is upregulated in AD mouse models and AD human brains.
ATP release from astrocytes in AD pathology may directly induce pathology. Conceivably, ATP activation of calcium-permeable pathways such as via P2X7 receptors or co-activation of NMDAR/P2X7 receptors could reduce the survival of neurons. On the other hand, a protective role of ATP released from astrocytes in AD has also been suggested.
6. Altered GABA Release from Astrocytes in AD
GABA is the major inhibitory neurotransmitter in the adult mammalian brain, and recent studies have also shown astrocytic release as a gliotransmitter in AD. In the healthy brain, cytosolic concentrations in astrocytes are kept at low levels. However, in human AD patients and mouse models of AD, GABA is aberrantly and abundantly produce and release via the anion channel bestrophin 1.
7. Altered D-Serine Release from Astrocytes in AD
NMDA receptors (NMDARs) play a central role in synaptic plasticity and learning, and memory. A distinctive feature of these receptors is the requirement for a co-agonist binding to the GluN1 subunit with glutamate binding to its recognition site on the GluN2-3 subunits for the activation of the receptor. While glycine was originally thought to be the co-agonist on NMDARs, several studies over the last few decades have demonstrated that D-serine is at least as important in modulating NMDARs.
D-serine is a putative gliotransmitter that has been linked to learning and memory by its actions on synaptic NMDARs. The astrocytic release of D-serine has been linked to the induction of LTP in the hippocampus and hypothalamus. Altered levels of D-serine have been associated with neurological disorders, including schizophrenia and epilepsy. Interestingly, D-serine levels in postmortem hippocampal and cortical samples of AD patients seem to be higher than in control patients.
Moreover, levels of both D-serine and serine racemase, the enzyme responsible for its synthesis, are also elevated in experimental models of AD. The increased levels of D-serine and NMDAR hyperfunction may contribute to memory impairments and excitotoxicity observed in AD.
8. Changes to Gliotransmission in Neurodegenerative Diseases Other Than AD
It is not unexpected that altered astrocyte–neuronal communication and gliotransmission are also found in neurodegenerative diseases other than AD. Indeed, impaired glutamate uptake by astrocytes is a well-documented dysfunction of astrocytes in Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD). Diminished levels of GLT-1 expression are found in postmortem brain tissues from patients with ALS, PD, or HD. As discussed above, dysregulation of expression and function of GLT-1 is linked to chronically high levels of extracellular glutamate causing excitotoxicity. Therefore, pharmacological induced upregulation of GLT-1 in neurodegenerative diseases is a promising approach to treating chronic excitotoxicity. Likewise, the enhanced expression of astrocytic Cx43 is altered in animal models of ALS and PD. As in AD, the high expression of Cx43 is hypothesized to cause excessive release of ATP or glutamate into the extracellular space, ultimately leading or contributing to neurodegeneration. Thus, there are many shared features in astrocyte–neuronal communication changes in diverse types of neurodegenerative diseases that may be a consequence of the astrocytic responses to their reactive state. Finding the commonalities and differences in astrocyte–neuronal communication in various neurodegenerative diseases may allow the design of novel strategies to treat these diseases.
9. Concluding Remarks