 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pilar Gonzalez-Cabo | + 2624 word(s) | 2624 | 2021-07-26 05:48:13 | | | |
| 2 | Lily Guo | Meta information modification | 2624 | 2021-07-29 04:23:23 | | |
Video Upload Options
Cofilin is an actin-binding protein that plays a major role in the regulation of actin dynamics, an essential cellular process. This protein has emerged as a crucial molecule for functions of the nervous system including motility and guidance of the neuronal growth cone, dendritic spine organization, axonal branching, and synaptic signalling. Recently, other important functions in cell biology such as apoptosis or the control of mitochondrial function have been attributed to cofilin. Moreover, novel mechanisms of cofilin function regulation have also been described. The activity of cofilin is controlled by complex regulatory mechanisms, with phosphorylation being the most important, since the addition of a phosphate group to cofilin renders it inactive. Due to its participation in a wide variety of key processes in the cell, cofilin has been related to a great variety of pathologies, among which neurodegenerative diseases have attracted great interest.
1. Introduction
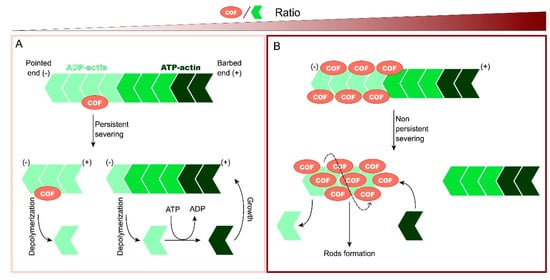
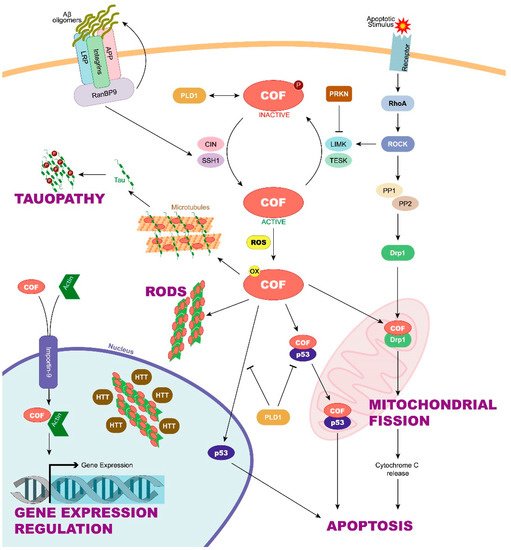
2. Cofilin Regulation and Its Implication in Neurodegenerative Diseases
3. New Functions for Cofilin in Neurodegenerative Diseases
4. Future Perspectives
References
- Bamburg, J.R.; Harris, H.E.; Weeds, A.G. Partial purification and characterization of an actin depolymerizing factor from brain. FEBS Lett. 1980, 121, 178–182.
- Bamburg, J.R. Proteins of the ADF/Cofilin Family: Essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 1999, 15, 185–230.
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin dynamics, architecture and mechanics in cell motility. Physiol. Rev. 2014, 94, 235–263.
- Bamburg, J.R.; Bernstein, B.W. Roles of ADF/cofilin in actin polymerization and beyond. F1000 Biol. Rep. 2010, 2, 62.
- Wioland, H.; Guichard, B.; Senju, Y.; Myram, S.; Lappalainen, P.; Jégou, A.; Romet-Lemonne, G. ADF/Cofilin accelerates actin dynamics by severing filaments and promoting their depolymerization at both ends. Curr. Biol. 2017, 27, 1956–1967.e7.
- Bernstein, B.W.; Chen, H.; Boyle, J.A.; Bamburg, J.R. Formation of actin-ADF/cofilin rods transiently retards decline of mitochondrial potential and ATP in stressed neurons. Am. J. Physiol. Cell Physiol. 2006, 291, C828–C839.
- Andrianantoandro, E.; Pollard, T.D. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/Cofilin. Mol. Cell 2006, 24, 13–23.
- Kanellos, G.; Frame, M.C. Cellular functions of the ADF/cofilin family at a glance. J. Cell Sci. 2016, 129, 3211–3218.
- Li, G.-B.; Zhang, H.-W.; Fu, R.-Q.; Hu, X.-Y.; Liu, L.; Li, Y.-N.; Liu, Y.-X.; Liu, X.; Hu, J.-J.; Deng, Q.; et al. Mitochondrial fission and mitophagy depend on cofilin-mediated actin depolymerization activity at the mitochondrial fission site. Oncogene 2018, 37, 1485–1502.
- Woo, J.-A.A.; Liu, T.; Fang, C.C.; Cazzaro, S.; Kee, T.; LePochat, P.; Yrigoin, K.; Penn, C.; Zhao, X.; Wang, X.; et al. Activated cofilin exacerbates tau pathology by impairing tau-mediated microtubule dynamics. Commun. Biol. 2019, 2, 112.
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71.
- Gibson, R.M. Does apoptosis have a role in neurodegeneration? BMJ 2001, 322, 1539–1540.
- McMurray, C.T. Neurodegeneration: Diseases of the cytoskeleton? Cell Death Differ. 2000, 7, 861–865.
- Mestre, H.; Hablitz, L.M.; Xavier, A.L.R.; Feng, W.; Zou, W.; Pu, T.; Monai, H.; Murlidharan, G.; Castellanos Rivera, R.M.; Simon, M.J.; et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. eLife 2018, 7, e40070.
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165.
- Verkhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiol. Rev. 2018, 98, 239–389.
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215.
- Alsegiani, A.; Shah, Z. The role of cofilin in age-related neuroinflammation. Neural Regen. Res. 2020, 15, 1451–1459.
- Kang, D.E.; Woo, J.A. Cofilin, a master node regulating cytoskeletal pathogenesis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 72, S131–S144.
- Zhang, Q.; Hu, C.; Huang, J.; Liu, W.; Lai, W.; Leng, F.; Tang, Q.; Liu, Y.; Wang, Q.; Zhou, M.; et al. ROCK1 induces dopaminergic nerve cell apoptosis via the activation of Drp1-mediated aberrant mitochondrial fission in Parkinson’s Disease. Exp. Mol. Med. 2019, 51, 1–13.
- Munsie, L.; Caron, N.; Atwal, R.S.; Marsden, I.; Wild, E.J.; Bamburg, J.R.; Tabrizi, S.J.; Truant, R. Mutant huntingtin causes defective actin remodeling during stress: Defining a new role for transglutaminase 2 in neurodegenerative disease. Hum. Mol. Genet. 2011, 20, 1937–1951.
- Ohashi, K. Roles of cofilin in development and its mechanisms of regulation. Dev. Growth Differ. 2015, 57, 275–290.
- Ohashi, K.; Nagata, K.; Maekawa, M.; Ishizaki, T.; Narumiya, S.; Mizuno, K. Rho-associated Kinase ROCK Activates LIM-kinase 1 by Phosphorylation at Threonine 508 within the Activation Loop. J. Biol. Chem. 2000, 275, 3577–3582.
- Van Troys, M.; Huyck, L.; Leyman, S.; Dhaese, S.; Vandekerkhove, J.; Ampe, C. Ins and outs of ADF/cofilin activity and regulation. Eur. J. Cell Biol. 2008, 87, 649–667.
- Huang, T.Y.; Minamide, L.S.; Bamburg, J.R.; Bokoch, G.M. Chronophin mediates an ATP-sensing mechanism for cofilin dephosphorylation and neuronal cofilin-actin rod formation. Dev. Cell 2008, 15, 691–703.
- Kim, J.-S.; Huang, T.Y.; Bokoch, G.M. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol. Biol. Cell 2009, 20, 2650–2660.
- Cameron, J.M.; Gabrielsen, M.; Chim, Y.H.; Munro, J.; McGhee, E.J.; Sumpton, D.; Eaton, P.; Anderson, K.I.; Yin, H.; Olson, M.F. Polarized cell motility induces hydrogen peroxide to inhibit cofilin via cysteine oxidation. Curr. Biol. 2015, 25, 1520–1525.
- Chua, B.T.; Volbracht, C.; Tan, K.O.; Li, R.; Yu, V.C.; Li, P. Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat. Cell Biol. 2003, 5, 1083–1089.
- Bernstein, B.W.; Shaw, A.E.; Minamide, L.S.; Pak, C.W.; Bamburg, J.R. Incorporation of cofilin into rods depends on disulfide intermolecular bonds: Implications for actin regulation and neurodegenerative disease. J. Neurosci. 2012, 32, 6670–6681.
- Zhao, H.; Hakala, M.; Lappalainen, P. ADF/cofilin binds phosphoinositides in a multivalent manner to act as a PIP(2)-density sensor. Biophys. J. 2010, 98, 2327–2336.
- Yeoh, S.; Pope, B.; Mannherz, H.G.; Weeds, A. Determining the differences in actin binding by human ADF and cofilin. J. Mol. Biol. 2002, 315, 911–925.
- Yoo, Y.; Ho, H.J.; Wang, C.; Guan, J.-L. Tyrosine phosphorylation of cofilin at Y68 by v-Src leads to its degradation through ubiquitin–proteasome pathway. Oncogene 2010, 29, 263–272.
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. 1), S32–S39.
- Huynh, D.P.; Scoles, D.R.; Ho, T.H.; Del Bigio, M.R.; Pulst, S.-M. Parkin is associated with actin filaments in neuronal and nonneural cells. Ann. Neurol. 2000, 48, 737–744.
- Lim, M.K.; Kawamura, T.; Ohsawa, Y.; Ohtsubo, M.; Asakawa, S.; Takayanagi, A.; Shimizu, N. Parkin interacts with LIM Kinase 1 and reduces its cofilin-phosphorylation activity via ubiquitination. Exp. Cell Res. 2007, 313, 2858–2874.
- Muñoz-Lasso, D.C.; Mollá, B.; Calap-Quintana, P.; García-Giménez, J.L.; Pallardo, F.V.; Palau, F.; Gonzalez-Cabo, P. Cofilin dysregulation alters actin turnover in frataxin-deficient neurons. Sci. Rep. 2020, 10, 5207.
- Barone, E.; Mosser, S.; Fraering, P.C. Inactivation of brain Cofilin-1 by age, Alzheimer’s disease and γ-secretase. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2500–2509.
- Kim, T.; Vidal, G.S.; Djurisic, M.; William, C.M.; Birnbaum, M.E.; Garcia, K.C.; Hyman, B.T.; Shatz, C.J. Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 2013, 341, 1399–1404.
- Pelucchi, S.; Vandermeulen, L.; Pizzamiglio, L.; Aksan, B.; Yan, J.; Konietzny, A.; Bonomi, E.; Borroni, B.; Padovani, A.; Rust, M.B.; et al. Cyclase-associated protein 2 dimerization regulates cofilin in synaptic plasticity and Alzheimer’s disease. Brain Commun. 2020, 2.
- Rehklau, K.; Hoffmann, L.; Gurniak, C.B.; Ott, M.; Witke, W.; Scorrano, L.; Culmsee, C.; Rust, M.B. Cofilin1-dependent actin dynamics control DRP1-mediated mitochondrial fission. Cell Death Dis. 2017, 8, e3063.
- Kanellos, G.; Zhou, J.; Patel, H.; Ridgway, R.A.; Huels, D.; Gurniak, C.B.; Sandilands, E.; Carragher, N.O.; Sansom, O.J.; Witke, W.; et al. ADF and cofilin1 control actin stress fibers, nuclear Integrity and cell survival. Cell Rep. 2015, 13, 1949–1964.
- Dopie, J.; Skarp, K.-P.; Kaisa Rajakylä, E.; Tanhuanpää, K.; Vartiainen, M.K. Active maintenance of nuclear actin by importin 9 supports transcription. Proc. Natl. Acad. Sci. USA 2012, 109, E544–E552.
- Han, L.; Stope, M.B.; de Jesús, M.L.; Oude Weernink, P.A.; Urban, M.; Wieland, T.; Rosskopf, D.; Mizuno, K.; Jakobs, K.H.; Schmidt, M. Direct stimulation of receptor-controlled phospholipase D1 by phospho-cofilin. EMBO J. 2007, 26, 4189–4202.
- Sun, Y.; Liang, L.; Dong, M.; Li, C.; Liu, Z.; Gao, H. Cofilin 2 in serum as a novel biomarker for Alzheimer’s disease in Han Chinese. Front. Aging Neurosci. 2019.
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput screening platforms in the discovery of novel drugs for neurodegenerative diseases. Bioengineering 2021, 8, 30.

