2. Cofilin Regulation and Its Implication in Neurodegenerative Diseases
Cofilin can undergo post-translational modifications that regulate its function and, among these, phosphorylation stands out. Phospho-regulation of cofilin is mediated by signalling pathways in response to extracellular signals. This signal transduction is very complex, and numerous studies have been carried out to characterize it [
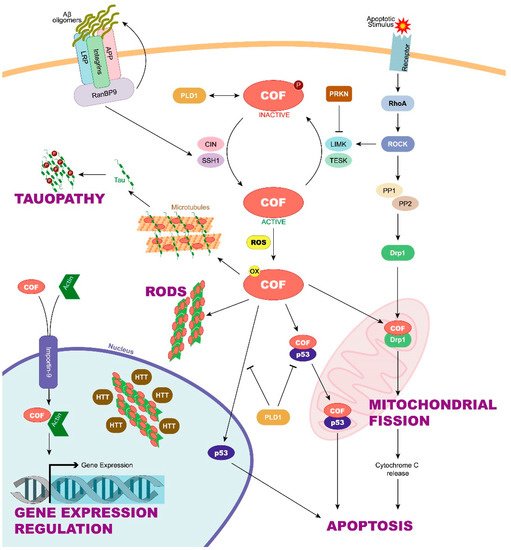
22]. Cofilin is inactive when it is phosphorylated on its Ser3 residue by LIM-domain containing kinases 1 and 2 (LIMK1 y LIMK2) and testis-specific kinases 1 and 2 (TESK1 y TESK2). LIMK1 is in turn activated by the small GTPase Rho and its downstream Rho-associated protein kinase (ROCK) [
23], and TESK1 is activated downstream of an integrin signal. Both contribute to stabilize the actin cytoskeleton. The phosphatases chronophin (CIN) and slingshot (SSH1) remove the phosphate of Ser3, activating cofilin and accelerating the dynamics of the actin cytoskeleton [
24]. However, in addition, the regulation of cofilin has to be spatiotemporally controlled to obtain a precise actin cytoskeleton reorganization. While cofilin is expressed in almost all tissues and cells, distinctly different expression patterns of LIMKs, TESKs, and SSHs exist [
22].
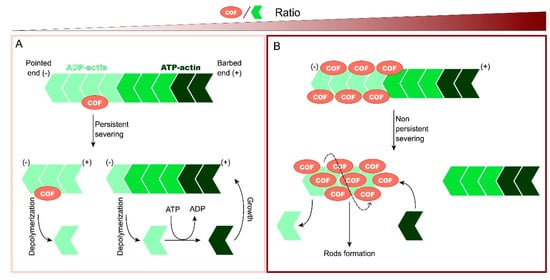
Moreover, other regulatory mechanisms of cofilin are known. Dephosphorylation by CIN and SSH is enhanced when ATP levels are low and reactive oxygen species (ROS) high [
25,
26], demonstrating how oxidative stress plays a role in the regulation of cofilin activity. Moreover, cofilin function is also directly regulated by the oxidation of its cysteine residues [
27]. Oxidized cofilin can induce the formation of an intramolecular disulfide bridge in vivo and the loss of its Ser3 phosphorylation site. This activated and oxidized cofilin is responsible for inducing the apoptosis cascade through its translocation to the mitochondria [
28]. In addition, cofilin oxidation can induce not only intramolecular but also intermolecular disulfide bonds between cofilin molecules. In these cases, cofilin–actin rod formation is enhanced, thus preventing the cell from entering apoptosis [
29]. Apart from phosphorylation and oxidation, other regulatory mechanisms have been proposed. For example, phosphatidylinositol 4,5-bisphosphate (PIP2) is a membrane phospholipid that inhibits cofilin activity by competing with actin at the binding site [
30]. High-pH conditions could also regulate the activity of cofilin by increasing it [
31]. Cofilin phosphorylation at its Y68 residue induces cofilin ubiquitination and degradation through the proteasome, adding another regulation mechanism [
32].
According to current evidence, it seems clear that cofilin is involved in neurodegenerative processes, as we will describe in the following paragraphs, and more specifically, the regulation of cofilin function has also been associated with different disease states. However, much remains to be known about the complex regulatory mechanisms of this protein and how they are affected in different neurodegenerative diseases.
Mutations in the Parkin 2 (
PARK) gene are the main cause of autosomal recessive parkinsonism, a type of early onset familial PD, and also seem to play a role in sporadic PD. Parkin is a ubiquitin E3 ligase that ubiquitinates dysfunctional proteins for their degradation in the proteasome. The loss of parkin activity leads to the accumulation of parkin substrates. These have been proposed as a cause for toxicity and neurodegeneration through mechanisms that are not completely understood [
33]. Previous studies had already shown that parkin was associated with actin filaments by colocalizing with them, so it was suggested that parkin could have a role in actin stabilization [
34]. According to this finding, Lim et al. observed that parkin interacts with and ubiquitinates LIMK1 in the human dopaminergic neuroblastoma cell line BE(2)-M17 but not in the human embryonic kidney-derived cell line HEK293, indicating a tissue-specific regulation. As a result, since LIMK1 phosphorylates cofilin, LIMK1 ubiquitination by PARK2 reduces the level of inactive cofilin [
35]. Overall, parkin modulates the level of phosphocofilin by negatively regulating LIMK1 activity in a cell-type-dependent way, which can help understand how cofilin contributes to familial PD.
Friedreich’s ataxia (FRDA) is an inherited peripheral neuropathy characterized by an early loss of neurons in the dorsal root ganglia, among other clinical symptoms caused by frataxin deficiency. Cytoskeletal abnormalities have been proposed to contribute to dying-back neurodegeneration in FRDA [
24]. In fact, at the molecular level, the F-actin:G-actin ratio has been shown to be increased in sensory neurons of FRDA mice, suggesting an alteration of the normal turnover of actin filaments. This disturbance could be the cause of the changes observed in the morphology of the growth cones of FRDA mouse neurons [
36]. The authors observed a hyperactivation of cofilin in the dorsal nerve roots of the FRDA mouse model compared to controls, which can be partially explained by the increased levels of CIN [
36]. Altogether, the dysregulation of cofilin might explain the reduced neurite growth and alterations of cytoskeleton, suggesting a role in FRDA neuropathy.
Regulation of active cofilin could also be involved in AD. A significant reduction of total SSH1 phosphatase in AD brains could be the cause of the increased cofilin inactivation observed in human samples. γ-Secretase is an enzyme that makes the second cleavage to amyloid precursor protein (APP) to give rise to the amyloid-β (Aβ) peptides, whose pathological accumulation is related to AD. This enzyme has been discovered to takes part in promoting actin/cofilin-related pathology. In this sense, γ-secretase inhibitors promote cofilin activation by increasing SSH1 levels in mouse primary cortical neurons [
37]. However, other authors point to the excessive cofilin activation, and not the opposite, as the cause of AD pathogenesis. For example, Kim et al. suggest that the binding of Aβ oligomers to the leukocyte immunoglobulin-like receptor B2 (LilrB2) present in human brain enhances cofilin signalling, which leads to synapse elimination [
38].
The contribution of active cofilin to AD pathogenesis does not seem to be limited only to its levels since its location also has an influence. Cofilin is translocated to the spine upon long-term potentiation induction and promotes the assembly of F-actin, essential for spine expansion and to potentiate synaptic transmission. This activity-dependent plasticity phenomenon is altered in the hippocampus of AD patients, with cofilin having a potential role in this defect since it is aberrantly localized in neuron spines [
39]. The authors showed how downregulation of cyclase-associated protein 2 (CAP2), by synergizing with cofilin to accelerate depolymerization of the pointed end of actin filaments, is able to control cofilin synaptic availability in long-term potentiation processes that, as it is known, are extremely important for stabilizing memory.
It seems clear that the regulation of cofilin is crucial for the activity and function of the protein, so that alterations in the cofilin regulation mechanisms trigger pathological processes such as neurodegenerative diseases.
3. New Functions for Cofilin in Neurodegenerative Diseases
Chua et al. have shown that after staurosporine-induced apoptosis, active cofilin can translocate from the cytoplasm to the mitochondria, leading to the opening of the mitochondrial permeability transition pore and the release of cytochrome C, which is the first step in the apoptotic cell death cascade [
28]. This suggests that cofilin has an important function during the initiation phase of apoptosis. In this regard, the translocation of cofilin to mitochondria has been also linked to mitochondrial fission, a process in which cofilin would participate regulating the function of the dynamin-related protein 1 (Drp1) [
40]. Moreover, cofilin could also be translocated to the nucleus due to its nuclear localization sequence [
41]. In this context, cofilin has been suggested to participate in actin import to the nucleus, where actin can regulate gene expression [
42]. Even inactive cofilin, a form that was thought to have no function, has been shown to have stimulatory effects on phospholipase D1 (PLD1), an enzyme involved in a wide variety of cellular responses, including calcium mobilization, superoxide production, endocytosis, vesicle trafficking, etc. [
43].
All these processes, highly related to neurodegeneration, point towards cofilin as a key protein involved in many neurological diseases, but its role in the pathophysiology of these diseases still remains to be elucidated.
4. Future Perspectives
Cofilin has a main role in actin dynamics and, therefore, in cytoskeletal homeostasis. However, current evidence on its new functions seems to show that cofilin also contributes to degenerative processes through the formation of cofilin–actin rods that impair axonal transport and promotion of neuronal cell death, or by changes in mitochondrial dynamics and in the endoplasmic reticulum–mitochondria (ER–mitochondria) connection and communication. However, much remains to be known about cofilin, starting with its complex regulatory mechanisms. This is a key piece to understand how the modulation of cofilin activity and levels could be a critical point in neurodegenerative diseases to modify its natural history. At this point, the question arises as to whether cofilin could be used as a neurodegenerative progression biomarker. In this sense, cofilin 2 expression was demonstrated to be significantly increased in the serum of Alzheimer’s disease patients, and it performed well as a diagnostic and non-invasive biomarker with high sensitivity and specificity [
72].
Thus, the biggest challenge is to determine whether cofilin is a potential target for neurodegenerative diseases, which can open new possibilities for drug development with the purpose, for example, of preventing mitochondrial alteration and axonopathy in neurodegenerative diseases. Another therapeutic approach could be to design drugs that target cofilin-regulatory proteins since many of the pathological processes related to cofilin are due to alterations in its regulation. Anyway, the recent advances in the use of high-throughput screening and computer-aided drug design can help in the search of treatments for neurodegenerative diseases [
73].