 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lucília Saraiva | + 3278 word(s) | 3278 | 2021-07-05 11:54:11 | | | |
| 2 | Dean Liu | + 652 word(s) | 3930 | 2021-07-29 03:10:59 | | |
Video Upload Options
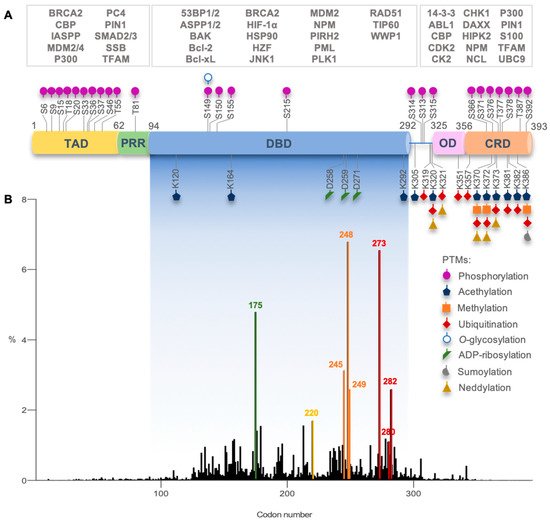
Active p53 is a homotetramer of four identical chains of 393 amino acids. Each monomer presents a modular structure divided into different domains: an acidic disorderedN-terminal region, comprising the transactivation domain (TAD) and the proline-rich region (PRR); a central core sequence-specific DNA-binding domain (DBD); aC-terminal region, encompassing an oligomerization domain (OD) and a disordered regulatory domain (CRD)().
1. Introduction
The p53 protein belongs to a family of transcription factors, alongside p63 and p73, whose full activity is essential for the prevention of tumor onset and development. It senses diverse physical or chemical distress cellular signals, such as DNA damage, that eventually lead to its activation, stabilization, and accumulation in the cell [1] Once active, p53 transcriptionally regulates many genes involved in major cellular processes, including cell cycle arrest, senescence, DNA damage repair, metabolic adaptation, and apoptosis [2] Importantly, p53 activity is tightly controlled by a complex feedback-regulated network involving endogenous negative regulators (e.g., Murine Double Minute 2, MDM2) as well as by post-translational modifications (PTMs) and by interaction with distinct signaling proteins [3][4][5]
The cell fate specified by p53 activation is context- and tissue-dependent, being mainly related to the nature of cellular stress [1]. In case of mild DNA damage, p53 drives cell cycle arrest for DNA repair, while under severe and irreparable DNA damage, p53 stimulates apoptotic or senescence programs. Other crucial factors, including the expression levels and PTM status of p53, cellular localization, co-factor enrollment, and the architecture of the promoters of target genes, also dictate the type of p53-mediated cellular responses based on the transcription of different subsets of p53 effectors [6][7].
The impairment of p53 tumor suppressor functions, either by mutations in theTP53gene (which encodes the p53 protein) or by other abnormalities in the p53 pathway, is a common event among human cancers. However, since its discovery, p53 has represented a challenging target for drug discovery, being frequently considered as “undruggable”. In fact, wild-type (wt) p53 and most p53 mutant (mutp53) forms lack binding pockets or allosteric sites, other than the DNA-binding groove itself, hindering rational drug design [8]. Nonetheless, in vitro and in vivo evidence has proven that restoration of mutp53, with re-establishment of wt-like activity, elicits impressive tumor regression and cell death [9][10][11][12].
2. p53 Structure and DNA Recognition
For this, specific recombinant p53 domains were produced by bioengineering techniques and analyzed by X-ray crystallography and NMR spectroscopy. In addition, some studies to check the spatial 3D arrangement of folded domains were pursued with the full-length protein, using suitable methodologies such as small angle X-ray scattering (SAXS), cryo-EM, and fluorescence resonance energy transfer (FRET) To complement these studies, electrophoretic mobility shift assay [13], protein microarrays, transactivation assays, fluorescence anisotropy, and isothermal calorimetric [13] titrations, among others, have also been used to unveil binding features and to characterize affinities for DNA and other proteins, peptides, or small molecules [14][15][16][17][18]. Recombinant p53 proteins with specific mutations (naturally occurring alone or combined with second-site mutations that can rescue DNA binding ability) have also contributed to the study of the relevance of specific amino acid residues in p53 stability and activity [19][20][21][22].
Active p53 is a homotetramer of four identical chains of 393 amino acids. Each monomer presents a modular structure divided into different domains: an acidic disorderedN-terminal region, comprising the transactivation domain (TAD) and the proline-rich region (PRR); a central core sequence-specific DNA-binding domain (DBD); aC-terminal region, encompassing an oligomerization domain (OD) and a disordered regulatory domain (CRD) [23] (Figure 1).
In solution, using SAXS, NMR, and EM techniques, it was possible to understand that full-length p53 forms a tetramer with an opened, cross-shaped structure, having the ODs at its center and a pair of loosely coupled DBD dimers at the ends, which are accessible to bind to cognate DNA and partner proteins. Upon DNA binding, the structure closes around DNA and becomes more compact [14][24][25] p53 is a biologically active transcription factor as a homotetramer (assembled as a dimer of dimers; p53 dimers are formed co-translationally [26]), and DNA recognition occurs upon a cooperative binding process [27].
p53 exerts its transcriptional activity through recognition of a short specific DNA sequence in the target genes’ promoters, called the response element (RE) [28][29]. Thus, upon stress stimuli, p53 can bind to multiple scattered REs in the genome, eliciting the transcription of sets of genes and related cellular responses [30]. In addition, mismatches from the consensus are frequent among established p53 REs at target gene promoters. Thus, there is a wide range in sequence-specific binding affinities, and this is correlated with variations in the protein-DNA contact geometry, providing insights into the mechanism of p53 function and regulation and the potential to elicit gene expression changes that are tailored to specific stress conditions through modulation of nuclear p53 protein levels or by specific PTMs or protein cofactors [28][31].
In 1994, Cho et al. reported for the first time the crystallographic model of p53 DBD with DNA at 2.2 Å [32]. The p53 DBD is an immunoglobulin-like centralβ-sandwich of two antiparallelβ-sheets, providing the basic scaffold for the DNA-binding surface (Figure 2B). This surface is constituted by two large loops, L2 and L3, the latter intercalated by a short helix, H1, stabilized by a tetrahedral coordination of two residues of each loop to a zinc atom (L2–C176, H179; L3–C238, C242) and by a loop-sheet-helix (LSH) motif constituted by L1,β-strands S2 and S2′, the end ofβ-strand S10 andC-terminal of H2 (Figure 2B).
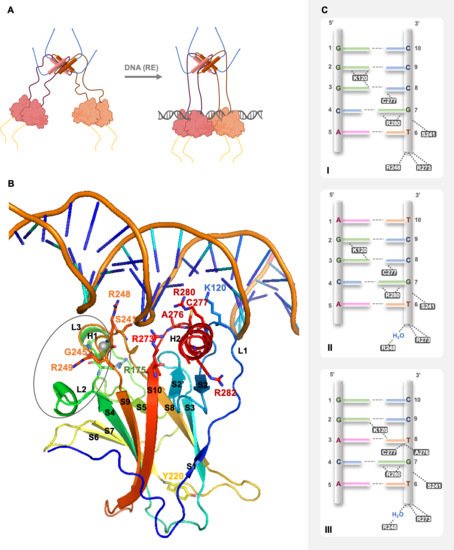
The LSH motif makes specific contacts with the major groove of the target DNA, through hydrogen bonds, salt-bridges, and structural water molecules in a tight network of hydrogen bonds that ensures the correct orientation of each amino acid residue. In the DNA major groove, the side chains of residues K120 (L1), A276, C277, and R280 (H2) interact with DNA bases via hydrogen bonds, and the side chain of residue R273 (S10) and the backbone amide of residues K120 and A276 establish interactions with the DNA backbone phosphates via salt bridge interactions (Figure 2B,C). The L3 contains the residues S241 and R248, which have contact with DNA backbone phosphates in the DNA minor groove; R248 is essential for DNA recognition (Figure 2B,C) [32][33].
Representation of p53 quaternary structure in solution, structural insights on p53 DBD binding to DNA, and p53 specific DNA-binding modes. The 3D model of the DBD binding to cognate DNA sequence Furthermore, it depicts the amino acid residues involved in DNA recognition, establishing interactions with DNA, specifically K120 (L1), S241 and R248 (L3), R273 (S10), A276, C277, and R280 (H2). The side chain of residue R280 is a steady point of contact between p53 and DNA, making two invariant hydrogen bonds to the conserved guanine base (G7) in the DNA major groove.
The interface is stabilized by hydrophobic, water-mediated polar interactions and hydrogen bonds between the zinc regions, involving H1, L2, and L3, of the two p53 DBDs (Figure 2B). There are two buried water molecules that provide an internal hydrogen-bonding network as a central anchor, linking the two zinc regions, which support H1 and L3 configuration. A nonpolar interaction shell is formed by surface residues from H1 and L3 hairpin via residues P177, H178, M243, and G244. Next to this hydrophobic area, there are two hydrophilic networks that contribute to its stability, one far from DNA, involving charged residues from the two monomers (specifically R174, E180, R181, and several water molecules), and the second one near to DNA, with a first-shell hydration of the protein surface facing DNA, via N239, S241, and R248 side chains [32].
The interaction of protein-DNA is the ultimate contribution to dimer stabilization. Dimer protein-protein interactions may also contribute to DBD integrity, considering the bidentated salt bridges between the polar and charged surface residues D184 with R175, and R249 with E171 on opposite sides of each monomer [28][34]. , in addition to acknowledging that both p53 DBD monomers in the dimer establish the same interactions with the DNA RE, advanced the hypothesis that, in a dimer, the p53 DBD interactions with DNA were stronger for one monomer than for the other [35]. In fact, this has also been supported by the spatial rearrangement of the tetramer complexed with DNA verified in cryo-EM works [14][24][25].
3. Mutant p53 Functions in Tumorigenesis
TP53mutations occur in more than half of human cancers, with colorectal, head and neck, esophagus, female genital organs, and lung cancers exhibiting the highest prevalence (37–43%; International Agency for Research on Cancer (IARC) [36].TP53mutations occur in both germline (associated with Li-Fraumeni syndrome) and sporadic contexts and can be found throughout the whole gene [36] (Figure 1B). In fact, in the p53 DBD, there are six hotspot missense mutations in codons 175, 245, 248, 249, 273, and 282, with high clinical significance [36] As previously mentioned, these codons code for residues with important roles in p53 structure and function.
TP53mutations in the TAD are associated with the loss of transactivation of specific genes, such asCDKN1A, hence blocking the capacity of inducing cell cycle arrest without compromising apoptosis. GOF can be manifested through mutp53 interaction with diverse transcriptional factors or co-factors. For instance, mutp53 heterooligomerizes with p63 or p73, blocking their tumor suppressor activity by inhibiting their transcriptional activity, or stimulating the transcription of non-canonical genes [37][38]. Both scenarios result in altered gene expression patterns that contribute to survival, tumor progression, and more aggressive phenotypes [38].
It is worth noting that mutp53 is expressed in cancer cells at higher levels than wtp53 in normal cells, indicating that mutp53 is somehow more stable than wtp53 and accumulates in tumor cells [39]. Nevertheless, MDM2 may be expressed by other pathways [40], and evidence has suggested that mutp53 levels can be controlled by MDM2 in normal tissues, but not in tumor tissues [41]. This raised the possibility of additional events in tumorigenesis responsible for mutp53 accumulation. Indeed, in cancer cells, the occurrence of mutp53 is also associated with increased levels of HSP70 or HSP90, which often bind to and stabilize mutp53 and/or participate in aggregosomes [42].
In general, p53 mutations lead to loss of DNA-binding ability and impairment of the p53 response (loss of function, LOF). (GOF) activities impact multiple hallmarks of cancer cell biology, affecting the chromatin structure, transcriptional regulation, and microRNA biogenesis, shaping the proteome, and rewiring tumor cell metabolic pathways. It also encompasses cytoplasmic functions and cell-extrinsic effects, namely affecting tumor microenvironment and the inflammatory response. Oncogenic GOF of mutp53, driving tumor development and dissemination, relies on the direct interaction with transcription factors (TFs, grey boxes) or co-factors and other protein effectors (yellow boxes), altering their activity, or on the transcriptional modulation of target genes (green boxes).
unveiled the deleterious effects of common cancer mutations [32][43]. In this context, based on the known p53 intrinsic instability, a superstable quadruple mutp53 DBD was developed (M133L, V120A, N239Y, N268D) and used for structural studies, enabling its handling without compromising its function [21][44]. The most frequentTP53mutations occur in highly conserved sequence regions and coincide with key amino acid residues for DNA recognition and structural stability Indeed, single amino acid mutations in the p53 DBD can result in the removal of DNA-contact residues (contact mutp53; e.g., R248Q, R248W, R273H, R273C, R280K) or conformational changes in different parts of the DBD, including structural rearrangements on the DNA-binding surface, creation of internal cavities, or formation of surface crevices in regions remote from the DNA-binding site (structural mutp53; e.g., G245S, G245D, R249S, R175H, Y220C, R282W) [43].
Mutp53 studies by X-ray crystallography and NMR have enlightened local structural changes and their impact on DNA recognition. In particular, NMR and in silico simulation data have provided insights into structural changes of L3, with L2 rearrangements, caused by the change of arginine to glutamine in the contact Conformational changes observed in the DNA-binding region of structural mutp53 G245S are small, with the overall conformation conserved [45]. This significant conformational change ultimately displaces R248, halting mutp53 DNA interaction [46].
Consequently, the L1 flexibility increases with displacement of the DNA-contact residue K120, affecting DNA recognition [47]. As the imidazole group of mutant histidine 220 roughly superimposes with the phenol of wt tyrosine, the interactions are partially conserved without observation of crevice formation when comparing with the other Y220 mutants [48]. Other mutations in theβ-sandwich may occur within its hydrophobic core, such as V143A and F270L. Valine and phenylalanine side chains form an integral part of the hydrophobic core network, and these mutations create internal cavities. Of note, these cavities occur without collapse of the surrounding structure guaranteed by other hydrophobic residues, but they cause a strong destabilization of the core domain [45].
Although no structural data of zinc region mutp53 R175H are available, it is possible to speculate why it abrogates p53 activity. If the arginine is replaced by a shorter and bulkier histidine (R175H), structural distortions and interference with zinc binding are expected [43]. In fact, for substitutions bearing smaller side chains, such in the case of R175A (not occurring in cancer), R175C, or R175L, residual binding [16] or wt-like activity [49] can be observed. Conversely, the introduction of large, bulky side chains (e.g., R175W and R175Y) abrogated the wt-like functions in cancer cells [49].
Impact of mutations on p53 DBD structural features and DNA recognition. Description of main physicochemical amino acid changes that influence the surrounding chemical environment, affecting interactions and structural rearrangements; consequences for mutp53 thermal stability, folding, and DNA-binding ability.
* The protein has four stabilizing mutations (M133L, V203A, N239Y, N268D).&Difference in free energy of unfolding between wt- and mutp53 DBD at 10 °C (ΔΔGD−NH2O), which is extrapolated to 37 °C; free energy of wtp53 used as reference (0 kcal/mol); (+) stable protein ≤ 0.5 kcal/mol, (+/−) weakly destabilized protein 0.6–2.5 kcal/mol, (−) strongly destabilized protein ≥ 2.6 kcal/mol.#Relative folded protein and DNA-binding affinity: (+++) 100–85%, (++) 84–70%, (+)
Regarding mutp53 stability, Bullock et al. have classified mutp53 DBD in accordance with its thermodynamic and kinetic stability and DNA-binding affinity, relating it to the mutation-specific local structural changes [15][16]. It was observed that mutp53 has higher propensity to aggregate than wtp53, with at least 50% of mutp53 denaturated at physiological temperature [16]. The structural distortion induced by mutp53 G245S and R249S slightly reduces protein thermal stability and halts DNA contacts, although G245S retains partial DNA-binding ability at sub-physiological conditions (20 °C). Similarly, in structural mutp53 Y220C or otherβ-sandwich mutants, despite the conservation of some DNA-binding affinity at 20 °C, the mutations dramatically decrease p53 stability with extensive protein unfolding at physiological temperature [16].
In addition, in the case of DNE, although there is a wtp53 dimer, the p53 DBD mutation in mutp53 dimer affects the overall tetramer stability, leading to the loss of binding cooperativity to DNA and subsequent impairment of transcriptional activity [26][27][43][50]. Moreover, mutations in the DBD not only affect DNA recognition, but may also interfere with binding to wtp53 target proteins. For instance, mutp53 forms bearing mutations that do not interfere with L2 and L3 conformation are still capable of interacting with TP53-binding protein 2 (53BP2); however mutp53 G245S impairs this interaction [43]. Importantly, mutp53 cellular half-life is not correlated with thermodynamic stability, as mutp53 tends to accumulate as previously mentioned [51][52].
Despite all the knowledge that has been generated around p53 structure and related topics, the elucidation of how several mutations impact on p53 conformation, stability, and function is still missing.
4. Targeting Mutant p53
Indeed, regarding mutp53, it could be arguable whether the reestablishment of its tumor suppressor function would be sufficient to counteract a context of multiple oncogenic alterations, including expression ofc-Myc, However, multiple in vitro and in vivo studies have proven the concept that the reactivation of wt-like function to mutp53 can elicit cell death and halt tumor progression [53][54]. Indeed, this provided the conceptual basis for the feasibility of mutp53 reactivation, by enhancing specific-sequence DNA-binding and transcription of wtp53 target genes. Another positive aspect of mutp53 as a therapeutic target is its high expression levels in cancer cells [55][56].
As formerly evidenced, p53 mutants are not all equal, differing on their structure, stability, and function. Therefore, reactivation of contact mutp53 seems to be a daunting task, since the therapeutic strategy must rely on the introduction of extra interaction points to compensate for the missing DNA contacts [57]. Conversely, structural mutants are found to be kinetically and thermodynamically destabilized in a temperature-dependent (e.g.,β-sandwich mutations) or temperature-independent (e.g., zinc region mutations) manner. For both structural mutant forms, small molecules acting as chaperones, increasing the level of correctly folded protein at physiological temperature, may be a feasible therapeutic strategy due to the conservation of the DNA-binding residues [16][57].
In the past two decades, consistent efforts from academic and industry research groups have led to the identification of several small molecules (and peptides) that stabilize p53 native conformation and restore sequence-specific DNA binding, rescuing wt-like transcriptional functions and ultimately resulting in cell death and tumor suppression (reviewed in [54][58]) (Table 2). Among these mutp53-targeting agents, two small molecule reactivators have entered clinical trials: PRIMA-1MET(APR-246; Phase III trials; NCT03745716 [59]) and COTI-2 (Phase I trial; NCT02433626 [60]).
Although, some mutp53-targeting agents have shown to bind and stabilize mutp53, the exact mechanism of mutp53 reactivation is still far from being fully understood [54]. The understanding of the precise molecular mechanism underlying the p53 activation of a mutp53-targeting agent, as well as of its off-target effects, is important data for the design of new, more efficient, and safe therapies.
This pocket is suitable to accommodate small molecules, thermodynamically stabilizing p53 in its native conformation and reactivating its transcriptional function [61]. A different approach targeting the DNA-binding surface of mutp53 In this regard, in silico results denoted a distinct molecular mechanism of mutp53 reactivation by SLMP53-1 and MANIO, not dependent on covalent bonds or mutation-created hydrophobic pockets, as described for other mutp53 reactivators. It is also interesting to note that these compounds can reactivate other mutp53 forms, both contact and structural, which further supports a molecular mechanism unrelated to the formation of pockets derived from specific mutation sites.
An alternative mutp53-targeting approach includes disruptors of protein-protein interaction, which inhibit the interaction of mutp53 with other proteins, thereby suppressing mutp53 The small molecule LEM2, besides inducing thermal stability to p73, also halted the interaction of mutp53 and p73, leading to tumor suppression by the transcription of p53-shared target genes [62]. Moreover, ReACp53, a mutp53 targeting peptide, has shown the capability to inhibit mutp53 containing aggregosomes by binding to S9 (an aggregation prone region) and favoring wt-like folding to structural mutp53 forms [63]. In silico studies disclosed that mutp53 forms exhibit an “open” state of the S6-S7 turn, exposing S9, and therefore representing a putative binding pocket to be explored in drug design [64].
Mutp53 protein degradation by small molecules, rather than its reactivation, with observed cellular growth inhibitory effect, constitutes another mutp53 targeting strategy [42]. This strategy could also be explored through a novel pharmacological approach, such as PROteolysis TArgeting Chimeras (PROTACs) [65]. For this, the combined knowledge of mutp53 structure and putative binding pockets or surfaces, known mutp53-targeting agents, and small molecules that bind to E3 ligases may boost the discovery of effective therapeutic options.
Although none of the reported mutp53-targeting compounds have reached the clinic, the structural and mechanistic diversity among them is encouraging. It is reasonable to expect that some of them will prove effective in clinical applications in the near future [54]. Furthermore, the pharmacological reactivation of mutp53 also poses a great opportunity in combination therapy with anticancer drugs known to trigger p53-dependent cancer cell death or to inhibit mutp53 downstream pathways [54].
However, it has to be considered that although the potential of reactivating wt-like functions of mutp53 with canonical gene transcription has been proven, this does not always translate into cell death or halt tumor growth, as this also depends on the cancer cell context [66][54].
References
- Riley, T.; Sontag, E.; Chen, P.A.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412.
- Menendez, D.; Inga, A.; Resnick, M. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737.
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582.
- Li, M.; He, Y.; Dubois, W.; Wu, X.; Shi, J.; Huang, J. Distinct Regulatory Mechanisms and Functions for p53-Activated and p53-Repressed DNA Damage Response Genes in Embryonic Stem Cells. Mol. Cell 2012, 46, 30–42.
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956.
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532.
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431.
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 1–13.
- Selivanova, G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014, 588, 2628–2638.
- Christophorou, M.A.; Martin-Zanca, D.; Soucek, L.; Lawlor, E.R.; Brown-Swigart, L.; Verschuren, E.; Evan, G.I. Temporal dissection of p53 function In Vitro and In Vivo. Nat. Genet. 2005, 37, 718–726.
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression In Vivo. Nat. Cell Biol. 2007, 445, 661–665.
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660.
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858.
- Tidow, H.; Melero, R.; Mylonas, E.; Freund, S.M.V.; Grossmann, J.G.; Carazo, J.M.; Svergun, D.I.; Valle, M.; Fersht, A.R. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc. Natl. Acad. Sci. USA 2007, 104, 12324–12329.
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.; Fersht, A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342.
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256.
- Lubin, D.J.; Butler, J.S.; Loh, S.N. Folding of Tetrameric p53: Oligomerization and Tumorigenic Mutations Induce Misfolding and Loss of Function. J. Mol. Biol. 2010, 395, 705–716.
- Tidow, H.; Veprintsev, D.; Freund, S.M.V.; Fersht, A.R. Effects of Oncogenic Mutations and DNA Response Elements on the Binding of p53 to p53-binding Protein 2 (53BP2). J. Biol. Chem. 2006, 281, 32526–32533.
- Ahn, J.-H.; Kim, T.J.; Lee, J.H.; Choi, J.-H. Mutant p53 stimulates cell invasion through an interaction with Rad21 in human ovarian cancer cells. Sci. Rep. 2017, 7, 1–11.
- Eldar, A.; Rozenberg, H.; Diskin-Posner, Y.; Rohs, R.; Shakked, Z. Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res. 2013, 41, 8748–8759.
- Nikolova, P.V.; Henckel, J.; Lane, D.; Fersht, A.R. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. USA 1998, 95, 14675–14680.
- Suad, O.; Rozenberg, H.; Brosh, R.; Diskin-Posner, Y.; Kessler, N.; Shimon, L.; Frolow, F.; Liran, A.; Rotter, V.; Shakked, Z. Structural Basis of Restoring Sequence-Specific DNA Binding and Transactivation to Mutant p53 by Suppressor Mutations. J. Mol. Biol. 2009, 385, 249–265.
- Joerger, A.C.; Fersht, A.R. The Tumor Suppressor p53: From Structures to Drug Discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919.
- Melero, R.; Rajagopalan, S.; Lázaro, M.; Joerger, A.; Brandt, T.; Veprintsev, D.; Lasso, G.; Gil, D.; Scheres, S.; Carazo, J.M.; et al. Electron microscopy studies on the quaternary structure of p53 reveal different binding modes for p53 tetramers in complex with DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 557–562.
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767.
- Nicholls, C.D.; McLure, K.G.; Shields, M.A.; Lee, P.W.K. Biogenesis of p53 Involves Cotranslational Dimerization of Monomers and Posttranslational Dimerization of Dimers. J. Biol. Chem. 2002, 277, 12937–12945.
- Weinberg, L.R.; Veprintsev, D.B.; Fersht, A.R. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004, 341, 1145–1159.
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural Basis of DNA Recognition by p53 Tetramers. Mol. Cell 2006, 22, 741–753.
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16.
- Tebaldi, T.; Zaccara, S.; Alessandrini, F.; Bisio, A.; Ciribilli, Y.; Inga, A. Whole-genome cartography of p53 response elements ranked on transactivation potential. BMC Genom. 2015, 16, 464.
- Jordan, J.J. Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet. 2008, 4, e1000104.
- Cho, Y.; Gorina, S.; Jeffrey, P.; Pavletich, N. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355.
- Wright, J.D.; Noskov, S.Y.; Lim, C. Factors governing loss and rescue of DNA binding upon single and double mutations in the p53 core domain. Nucleic Acids Res. 2002, 30, 1563–1574.
- Ho, W.C.; Fitzgerald, M.X.; Marmorstein, R. Structure of the p53 Core Domain Dimer Bound to DNA. J. Biol. Chem. 2006, 281, 20494–20502.
- Kantarci, N.; Doruker, P.; Haliloglu, T. Cooperative Fluctuations Point to the Dimerization Interface of P53 Core Domain. Biophys. J. 2006, 91, 421–432.
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876.
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168.
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286.
- Prives, C.; White, E. Does control of mutant p53 by Mdm2 complicate cancer therapy? Genes Dev. 2008, 22, 1259–1264.
- Manfredi, J.J. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010, 24, 1580–1589.
- Lang, G.A.; Iwakuma, T.; Suh, Y.-A.; Liu, G.; Rao, V.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell 2004, 119, 861–872.
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606.
- Joerger, A.C.; Fersht, A.R. Structure–function–rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242.
- Joerger, A.; Allen, M.D.; Fersht, A.R. Crystal Structure of a Superstable Mutant of Human p53 Core Domain. J. Biol. Chem. 2004, 279, 1291–1296.
- Joerger, A.; Ang, H.C.; Fersht, A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061.
- Joerger, A.; Ang, H.C.; Veprintsev, D.; Blair, C.M.; Fersht, A.R. Structures of p53 Cancer Mutants and Mechanism of Rescue by Second-site Suppressor Mutations. J. Biol. Chem. 2005, 280, 16030–16037.
- Zhang, Y.; Coillie, S.V.; Fang, J.-Y.; Xu, J. Gain of function of mutant p53: R282W on the peak? Oncogenesis 2016, 5, e196.
- Bauer, M.R.; Krämer, A.; Settanni, G.; Jones, R.N.; Ni, X.; Tareque, R.K.; Fersht, A.R.; Spencer, J.; Joerger, A.C. Targeting Cavity-Creating p53 Cancer Mutations with Small-Molecule Stabilizers: The Y220X Paradigm. ACS Chem. Biol. 2020, 15, 657–668.
- Ryan, K.M.; Vousden, K.H. Characterization of Structural p53 Mutants Which Show Selective Defects in Apoptosis but Not Cell Cycle Arrest. Mol. Cell. Biol. 1998, 18, 3692–3698.
- Aramayo, R.; Sherman, M.B.; Brownless, K.; Lurz, R.; Okorokov, A.; Orlova, E.V. Quaternary structure of the specific p53–DNA complex reveals the mechanism of p53 mutant dominance. Nucleic Acids Res. 2011, 39, 8960–8971.
- Wawrzynow, B.; Zylicz, A.; Zylicz, M. Chaperoning the guardian of the genome. The two-faced role of molecular chaperones in p53 tumor suppressor action. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1869, 161–174.
- Yamamoto, S.; Iwakuma, T. Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers 2018, 11, 4.
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370.
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102.
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713.
- Terzian, T.; Suh, Y.-A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008, 22, 1337–1344.
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404.
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288.
- NIH-ClinicalTrials. APR-246 & Azacitidine for the Treatment of TP53 Mutant Myelodysplastic Syndromes (MDS). 2018. Available online: (accessed on 11 April 2021).
- NIH-ClinicalTrials. Study of COTI-2 as Monotherapy or Combination Therapy for the Treatment of Malignancies (COTI2-101). 2019. Available online: (accessed on 11 April 2019).
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.-K.; Hall, L.V.; Salehi, F.; Lin, D.-W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407.
- Gomes, S.; Raimundo, L.; Soares, J.; Loureiro, J.B.; Leão, M.; Ramos, H.; Monteiro, M.N.; Lemos, A.; Moreira, J.; Pinto, M.; et al. New inhibitor of the TAp73 interaction with MDM2 and mutant p53 with promising antitumor activity against neuroblastoma. Cancer Lett. 2019, 446, 90–102.
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Nguyen, T.Q.A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103.
- Pradhan, M.R.; Siau, J.W.; Kannan, S.; Nguyen, M.N.; Ouaray, Z.; Kwoh, C.K.; Lane, D.P.; Ghadessy, F.; Verma, C.S. Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic Acids Res. 2019, 47, 1637–1652.
- Demir, Ö.; Barros, E.P.; Offutt, T.L.; Rosenfeld, M.; Amaro, R.E. An integrated view of p53 dynamics, function, and reactivation. Curr. Opin. Struct. Biol. 2021, 67, 187–194.
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078.



