Cancer cells alter metabolic processes to sustain their characteristic uncontrolled growth and proliferation. Altered metabolic flux in cancer is controlled by tumor-host cell interactions, key oncogenes, tumor suppressors, and other regulatory molecules, including non-coding RNAs. Changes to metabolic pathways in cancer are dynamic, exhibit plasticity, and are often dependent on the type of tumor and the tumor microenvironment, leading in a shift of thought from the Warburg Effect and the “reverse Warburg Effect” to metabolic plasticity.
1. Introduction
Cancer is a complex genetic disease that arises from elaborate changes to the genome. This includes a cumulative collection of gain-of-function mutations that stimulate oncogenes, loss-of-function mutations that inactivate tumor suppressor genes, and mutations that inactivate stability genes involved in proliferative cell division, all of which help facilitate the transformation of a cell to a malignant phenotype. Characteristics typical of cell with a malignant phenotype include an unlimited ability to replicate, avoidance of apoptosis, insensitivity to anti-growth signals, continuous angiogenesis, self-sustained growth signals, tissue invasion, and metastasis [1]. In order for malignant cells to obtain the energy and materials required for acquisition and maintenance of these characteristics, they must undergo reprogramming of their metabolic pathways. Enhanced growth and proliferation via replicative division in cancer cells means they also require an increased amount of energy in the form of ATP, co-factors, such as NADPH and NADH, and building block molecules, such as carbon skeletons, to assemble new daughter cells. The increased demand for materials is satisfied through alteration of flux through key cellular metabolic pathways. Glycolysis and glucose metabolism are the most well-known altered metabolic pathways in cancer cells, with the first observations made about 100 years ago. Since then, there have been many other pathways found to be altered in cancer, such as glutamine metabolism, lipid acquisition, fatty acid oxidation, one-carbon metabolism, branched chain amino acid metabolism, and the citric acid cycle. The reprogramming of these pathways involves complex mechanisms and the coordination of a variety of signaling molecules, including molecules previously regarded as insignificant, non-coding RNAs. Furthermore, because of the complexity of these mechanisms, the reprogramming of metabolic pathways in cancer often occurs in various degrees and contexts in many types of cancer, affording cancer cells a plasticity that is not observed in normal cells. Much progress has been made in understanding the intricate nature of these pathways, providing a solid foundation for the development of new cancer therapies.
2. Glucose Metabolism and the Warburg Effect: A Century Later
Otto Warburg’s description of glucose metabolism in cancer cells was made almost a century ago, and it remains a key concept in the field of cancer metabolism. The Warburg Effect states that cancer cells rely on aerobic glycolysis (the conversion of glucose to lactate in the presence of oxygen) for ATP production, as compared to normal cells that rely on oxidative phosphorylation (OXPHOS) [2].
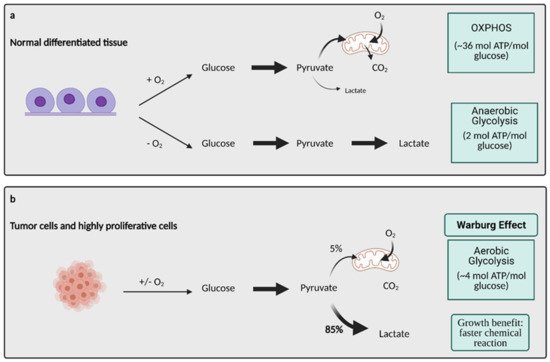
Glucose metabolism is considered one of the most important aspects of cancer cell metabolism as it supplies intermediates and precursors for several other key metabolic pathways, including the generation of amino acids, nucleotides, and lipids [3]. Thus, one of the first questions posed about glucose metabolism in cancer is why cells would shift towards an energy generation mechanism that is less efficient. OXPHOS generates 36 ATPs per molecule of glucose while glycolysis only generates 2 ATPs for one glucose molecule [3]. Lactate generation, however, is an approximately two order of magnitude faster chemical reaction than OXPHOS, and therefore, offers a growth benefit (Figure 1) [4]. In order to obtain the uncontrolled growth that is characteristic of cancer, cells need to generate energy quickly, and this is accomplished effectively with aerobic glycolysis. The high output of lactate also generates an acidic microenvironment where only cells with phenotypes resistant to acidic environments can grow. This offers a huge growth advantage and intensifies the invasive and metastatic nature of cancer cells, as other cells around them deteriorate [5].
Figure 1. Glucose metabolism in normal differentiated tissue vs. tumor cells. (
a) In normal differentiated tissues, one of two pathways is utilized. When oxygen is present, glucose is metabolized to pyruvate, which later enters OXPHOS to produce ~36 mol ATP/mol glucose. When no oxygen is present, glucose is metabolized to lactate, which yields 2 mol ATP/mol glucose. (
b) Tumors and other highly proliferative cells prefer to convert the majority of their glucose to lactate to yield ~4 mol ATP/mol glucose, even in the presence of oxygen. This is called the Warburg Effect, and while it produces less ATP/mol glucose, it is a much faster chemical reaction than OXPHOS, conferring a major growth benefit to cancer cells. ATP; adenosine triphosphate, CO
2; carbon dioxide, O
2; oxygen, mol; mole. Figure created with
BioRender.com (accessed on 26 March 2021).
As hypoxia increases in the tumor microenvironment, the cells employ certain stress responses as a means of survival
[6]. These stress responses are mediated by oncogenes and tumor suppressors that activate specific molecules and signaling pathways that are key in regulating aerobic glycolysis. Some examples of these regulators are proto-oncogene Myc, transcription factor hypoxia inducible factor 1 (HIF-1), the PI3K/Akt/mTOR pathway, and tumor suppressor p53, all which are known to be expressed abnormally or altered in many different types of cancers
[7].
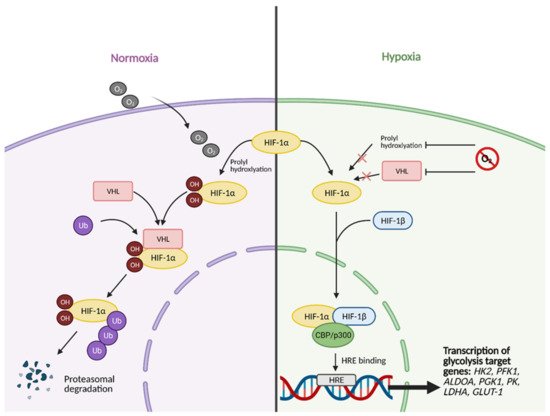
All of the aforementioned pathways and molecules have crosstalk with the master regulator and oxygen-sensing transcription factor HIF-1
[8]. Some examples include the activation of regulatory subunit HIF-1α by Akt and mTOR
[9], the inhibition of Myc by HIF under hypoxic conditions and cooperation between the two molecules to promote cancer cell growth
[10], and the inhibition of HIF-1α via high p53 expression
[11]. This molecular crosstalk is especially important because HIF-1 exerts major effects on glucose metabolism when active. Unstable in highly oxygenic conditions, the regulatory subunit of HIF-1, HIF-1α, becomes stabilized under hypoxic conditions, allowing it to translocate to the nucleus and form a heterodimer with its binding partner, HIF-1β (
Figure 2)
[7].
Figure 2. HIF-1α activation in normal vs. hypoxic conditions. Under normal conditions, in the presence of oxygen, the regulatory subunit of master transcription factor hypoxia-inducible factor (HIF-1), HIF-1α, undergoes prolyl hydroxylation, which induces HIF-1α binding to von Hippel-Lindau (VHL) tumor suppressor protein. This results in HIF-1α being tagged with Ubiquitin (Ub) to undergo proteasomal degradation. In hypoxic conditions, HIF-1α cannot undergo prolyl hydroxylation and subsequent binding to VHL. This allows it to bind to its partner, HIF-1β, and translocate to the nucleus. In the nucleus, fully functional HIF then forms a complex with transcriptional co-activators CBP/p300. The complex then binds to hypoxia response element (HRE) domains of the DNA, resulting in the transcription of several genes involved in glycolysis, such as HK2 (hexokinase 2), PFK1 (phosphofructokinase 1), ALDOA (aldolase A), PGK1 (phosphoglycerate kinase 1), PK (pyruvate kinase), LDHA (lactate dehydrogenase A), and GLUT-1 (glucose transporter 1). O2; oxygen, OH; hydroxyl group. Figure created with
BioRender.com (accessed on 26 March 2021).
Recently, a “metabolic plasticity” theory of cancer cells has been described, where cells still have fully functional OXPHOS machinery and can switch between OXPHOS and aerobic glycolysis, or even perform them simultaneously
[8]. This affords them the ability to adapt to various microenvironments and provides a mechanism of chemoresistance.
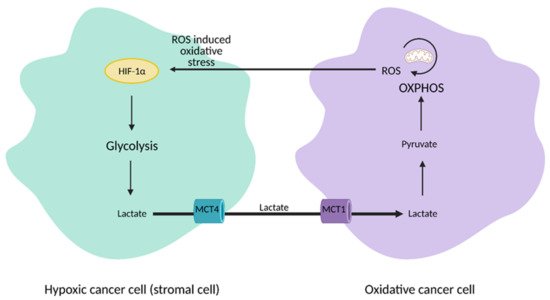
The discovery of heterogeneity in tumors led to a paradigm shift from the Warburg Effect to the “reverse Warburg Effect”, where aerobic glycolysis in cancer cells metabolically supports adjacent cancer cells (
Figure 3). This allows for the transfer of metabolites, such as lactate, to these cells to encourage ATP production, growth, and proliferation via oxidative phosphorylation
[12][13].
Figure 3. Reverse Warburg Effect. In the reverse Warburg Effect, substrates from different populations of cancer cells can be shared between each other and utilized. Oxidative cancer cells can take up lactate from hypoxic cancer cells that perform aerobic glycolysis to fuel oxidative phosphorylation (OXPHOS). Hypoxic cancer cells can also take up reactive oxygen species (ROS) from oxidative cancer cells to induce hypoxia-inducible factor 1α (HIF-1α) activation and aerobic glycolysis. This affords cancer cells an additional mechanism that enhances proliferation and survival. MCT1; monocarboxylate transporter 1, MCT4; monocarboxylate transporter 4. Figure created with
BioRender.com (accessed on 26 March 2021).
3. Glutamine Metabolism
A second key source of energy for cancer cells is the essential amino acid glutamine (Gln). Glutamine is the most consumed amino acid in cancer and the dependence on glutamine for growth is a hallmark of the disease [14]. Cancer cells deprived of glutamine rapidly die off [15], and increased glutamine metabolism in cancer is often referred to as “glutamine addiction”.
To attenuate oxidative damage and produce additional macromolecules, cancer cells increase the process of glutaminolysis, although the levels vary with heterogeneity of tumor, patient, and cancer type [16].
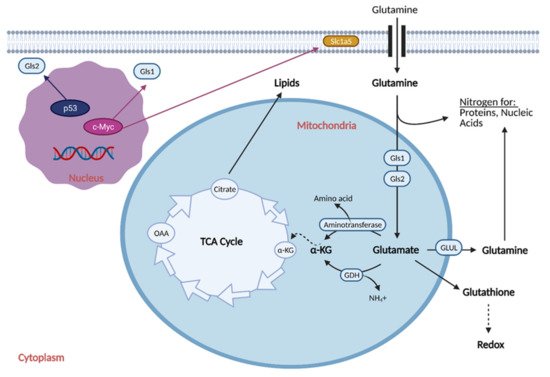
Reprogramming the cell to perform glutaminolysis is achieved through various oncogenes and is thought to be coordinated with the reprogramming of glucose metabolism (Figure 4) [17].
Figure 4. Glutamine Metabolic Reprogramming in Cancer. Cancer cells exhibit increased glutaminolysis, which is the conversion of glutamine to glutamate. This occurs as a result of upregulation of an isoform of glutaminase (Gls1) and glutamine transporter, Slc1a5, by oncogenic c-Myc. Increased glutamine uptake provides nitrogen for proteins and nucleic acids, while increased glutaminolysis provides α-ketoglutarate (α-KG) for the citric acid (TCA) cycle, resulting in increased production of lipids. Increased glutamine uptake also results in the production of glutathione, which regulates redox and helps the cell attenuate oxidative damage. Cancer cells typically exhibit downregulation of a second isoform of glutaminase, Gls2, as induction of its expression by p53 generally leads to tumor suppression. GDH; glutamate dehydrogenase, GLUL; glutamine synthetase, NH4+; ammonium, OAA; oxaloacetate. Figure created with
BioRender.com (accessed on 26 March 2021).
Recently, research has described a shift in glutamine nitrogen metabolism, referred to as a “second Warburg-like effect”
[18]. This effect describes a change in metabolism in cancer cells from glutaminolysis to de novo nucleotide biosynthesis. Although glutaminolysis has long been considered to be a tumor promoting factor, recent evidence has demonstrated that glutaminolysis may restrict nucleotide biosynthesis and impair cancer cell proliferation.
4. Pentose Phosphate Pathway
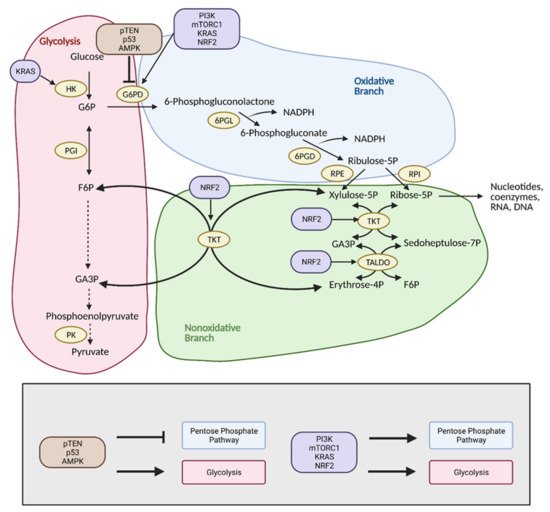
The pentose phosphate pathway (PPP), also referred to as the hexose monophosphate shunt, is an offshoot pathway of glycolysis that plays an important role in glucose metabolism, and consequently, cancer metabolism
[19]. Although focus has been heavily placed on increased glycolytic flux in cancer, recent research shows that cancer cells may metabolically reprogram themselves to direct glucose flux into the PPP
[20]. After glucose is converted into glucose-6-phosphate (G6P) by hexokinase (HK), it can be further metabolized by glycolysis. Alternatively, G6P can enter the PPP, which serves to generate NADPH and precursors to both lipids and nucleotides. These molecules encourage tumor growth by providing cells with the energy and substrates necessary for the synthesis of macromolecules
[20]. With the production of NADPH, the PPP also provides increased antioxidant defense for cancer cells in stress conditions to ensure their survival and proliferation, and thus, the reason why tumors tend to exhibit increased flux into this pathway (
Figure 5)
[21].
Figure 5. Reprogramming of the pentose phosphate pathway (PPP) in cancer. Upregulation of the PPP is achieved primarily through upregulation of glucose-6-phosphate dehydrogenase (G6PD) by PI3K, mTORC1, KRAS, and NRF2. This shunts glucose into the oxidative branch of the PPP instead of into glycolysis. Inhibitors of G6PD, pTEN, p53, and AMPK are often found mutated in cancer. Upregulation of the nonoxidative branch of the PPP in cancer occurs via NRF2, which increases transketolase (TKT) and transaldolase (TALDO) expression. There is also crosstalk between glycolysis and the PPP via KRAS, which increases hexokinase (HK) expression to upregulate glycolytic intermediates for progression into the PPP. Modulation of the PPP in this manner results in the production of energy and substrates necessary for tumor growth. F6P; fructose 6-phosphate, G3P; glyceraldehyde 3-phosphate, G6P; glucose 6-phosphate, NADPH; nicotinamide adenine dinucleotide phosphate, 6PGD; 6-phosphogluconate dehydrogenase, 6PGL; 6-phosphogluconolactonase, PK; pyruvate kinase, PGI; phosphoglucoisomerase, RPE; ribulose 5-phosphate 3-epimerase, RPI; ribose-5-phosphate isomerase. Figure created with
BioRender.com (accessed on 26 March 2021).
5. Lipid Metabolism
Aberrant lipid metabolism is one of the most pronounced metabolic alterations in cancer, and it greatly contributes to cancer cell growth and tumorigenesis
[22]. Lipids, including sterols, mono/di/triglycerides, phospholipids, and glycolipids, are indispensable to cells. They serve as energy sources, as components of biological membranes, and as signaling molecules
[23]. The many roles of lipids are a testament to the importance of processes that regulate their levels in cancer. Several aspects of lipid metabolism are reprogrammed in cancer, including the biosynthesis and oxidation of fatty acids (FAs), the uptake of FAs from the environment, and modification of FAs and release from other molecules (
Figure 6)
[22], of which the mechanisms will be discussed.
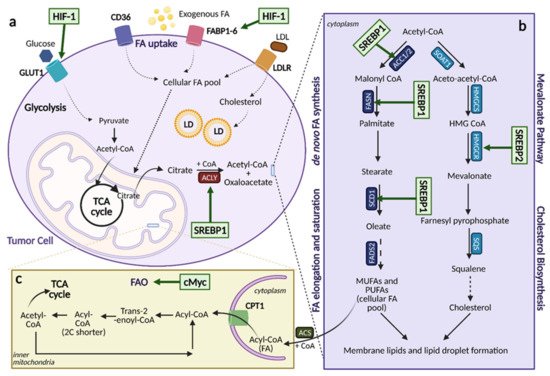
Figure 6. Lipid Metabolic Reprogramming in Cancer. An overview of lipid metabolic pathways and how they are modified in cancer. (
a). Tumor cells take up fatty acids (FAs) using multiple transporters, including CD36, FA binding proteins 1-6 (FABP1-6), and low-density lipoprotein receptor (LDLR) for low-density lipoproteins (LDL). These free FAs then become a part of the cellular FA pool where they can enter the citric acid (TCA) cycle and contribute to lipid formation. Upregulation of FA uptake in cancer occurs through hypoxia-inducible factor (HIF-1)-induced FABP1-6 overexpression. (
b). Upregulation of lipogenesis and cholesterol biosynthesis is achieved through sterol regulatory element binding protein (SREBP) activation. SREBP1 activation induces expression of lipogenesis genes while SREBP2 activation induces expression of cholesterol biosynthesis genes. (
c). Fatty acid oxidation (FAO) can be upregulated by cMyc, depending on the cancer type as a means to counteract oxidative stress. ACC1/2; acetyl-CoA carboxylase 1/2, ACLY; ATP citrate lyase, ACS; acyl-CoA synthetase, α-KG; alpha-ketoglutarate, CoA; coenzyme A, CPT1; carnitine palmitoyltransferase 1, FADS; FA desaturases, FASN; fatty acid synthase, FPP; farnesyl-pyrophosphate, GLUT1; glucose transporter 1, HMG-CoA; hydroxy-methylglutaryl-CoA, HMGCS; hydroxy-methylglutaryl-CoA synthase, HMGCR; hydroxy-methylglutaryl-CoA reductase, LD; lipid droplets, MUFA; monounsaturated fatty acids, PUFA; polyunsaturated fatty acids, SCD1; stearoyl-CoA desaturase 1, SOAT; sterol O-acyltransferase. Figure created with
BioRender.com (accessed on 26 March 2021).
6. The Tricarboxylic Acid (TCA) Cycle
It was previously thought that cancer cells bypass the TCA cycle and favor aerobic glycolysis. Recent evidence, however, suggests that cancer cells do rely heavily on the TCA cycle for energy production and growth [24]. This is achieved through the uncoupling of glycolysis from the TCA cycle, which allows for the use of alternate fuel sources to support increased metabolic demands [24].
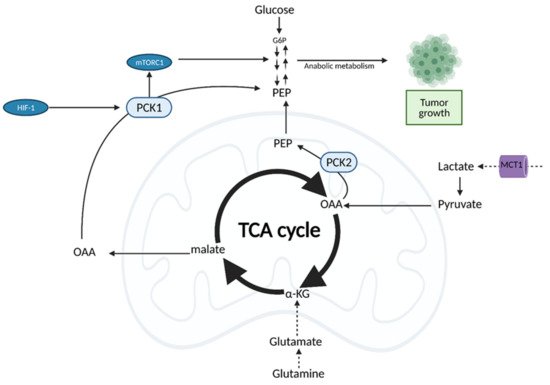
Both normal cells and tumor cells can catabolize every type of fuel that feeds the TCA cycle, including glucose, glutamine, and fatty acids; however, they differ in the rate of utilization and uptake of each fuel. While normal cells primarily use the conversion of glucose to pyruvate to fuel the TCA cycle, cancer cells typically shunt glucose away from the TCA cycle for breakdown in aerobic glycolysis. As a result, cancer cells are more dependent on glutamine and fatty acids to fuel the TCA cycle (Figure 7), although the exact levels of substrate utilization vary based on cancer type [24].
Figure 7. Oncogenic Regulation of the citric acid (TCA) cycle to support tumor growth. Unlike normal cells which primarily utilize glucose for input into the TCA cycle, cancer cells rely on alternative substrates, such as glutamate produced via glutaminolysis and lactate. TCA cycle flux is modulated by phosphoenolpyruvate carboxykinase (PEPCK), which has both cytosolic and mitochondrial isoforms (PCK1/2). PEPCK is overexpressed via hypoxia-inducible factor (HIF-1) and preferentially uses OAA derived from lactate as a substrate. Increased anaplerosis into the TCA cycle is compensated by the cataplerotic conversion of oxaloacetate (OAA) to phosphoenolpyruvate (PEP) via PEPCK. Overexpression of PEPCK promotes cancer cell growth via a truncated form of gluconeogenesis to glycolytic intermediates. These intermediates can be used for anabolic metabolism to support tumor growth. α-KG; alpha-ketoglutarate, G6P; glucose 6-phosphate, MCT1; monocarboxylate transporter 1. Figure created with
BioRender.com (accessed on 26 March 2021).
Importantly, cancer cells can also use other substrates for the TCA cycle, such as lactate. Although lactate was primarily considered a byproduct of aerobic glycolysis in the tumor environment, recent studies show that pancreatic, breast, and lung cancers utilize lactate for the TCA cycle, and even preferentially, over glucose
[25][26][27].
There are also several mutations in TCA cycle enzymes associated with tumor proliferation, including mutations in aconitase (ACO2), citrate synthase (CS), succinate dehydrogenase (SDH), fumarate hydratase (FH), and isocitrate dehydrogenase (IDH)
[27]. Notably, SDH and IDH mutations lead to increased production of ROS and promote tumorigenesis while FH mutations lead to the accumulation of fumarate, which can act as oncometabolite and allows for HIF stabilization
[27][28][29].
7. Acetate
Certain cancer cells may have an “acetate addiction” similar to the well-studied “glutamine addiction”, as the proliferation of normal cells is not affected by a lack of ACSS2 [30].
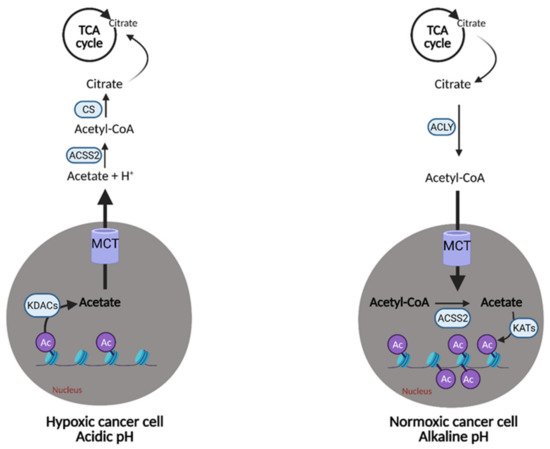
Acetate also represents another route of “metabolic plasticity” for cancer cells. In hypoxic conditions, when OXPHOS is compromised, or when the availability of exogenous FAs is low, acetate can be converted to acetyl CoA for use in the TCA cycle or production of biomass. The role of acetate in histone acetylation can be exploited by cancerous cells as an adaption and growth mechanism. Lysine acetyltransferases (KATs) catalyze the transfer of the acetyl group from acetyl-CoA for acetylation, and conversely, lysine deacetylases (KDACs) containing zinc (ZnKDACs) catalyze the deacetylation of histones, which releases free acetate. This free acetate can then be exported or converted back to acetyl CoA via acetyl CoA synthetases. Zn-KDACs are often over-expressed in various cancers, resulting in an increased release of free acetate from the cells, providing a mechanism of pH adaptation in cancer cells. In the acidic tumor microenvironment, the cell can release free acetate to buffer itself and alleviate metabolic stress for a short period of time (
Figure 8), thus, supporting survival and growth until other metabolic pathways can catch up and compensate
[31].
Figure 8. Metabolic plasticity of acetate in cancer. Cancer cells can alter the concentration of free acetate based on conditions in the tumor microenvironment. In hypoxic/acidic conditions, when oxidative phosphorylation (OXPHOS) is compromised or when there is low availability of exogenous fatty acids (FAs), cancer cells can release free acetate to raise the pH inside of the cell or convert it into acetyl-CoA for use in the citric acid (TCA) cycle. This is accomplished via the release of acetate from acetylated histones in the nucleus by lysine deacetylases (KDACs). Alternatively, cancer cells with functional OXPHOS or an excess of free FAs, can uptake free acetate via the acetylation of histones in the nucleus by lysine acetyltransferases (KATs). This buffering system provides cancer cells with another survival and growth mechanism. Ac; acetyl group, Acetyl-CoA; acetyl-coenzyme A, ACLY; adenosine triphosphate (ATP) citrate lyase, ACSS2; acetyl-CoA synthetase 2, CS; citrate synthase, H+; hydrogen, MCT; monocarboxylate transporter. Figure created with
BioRender.com (accessed on 26 March 2021).
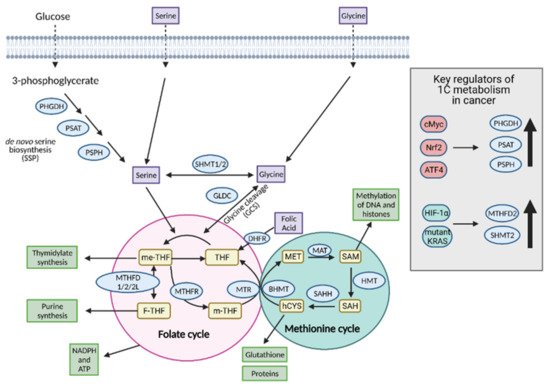
8. One-Carbon Metabolism
One-carbon (1C) metabolism involves the transfer and cycling of 1C-groups between various acceptor groups for biosynthesis
[32]. 1C metabolism controls the synthesis of purines, thymidine, glutathione, and S-adenosylmethionine (SAM), which are ultimately converted to proteins, lipids, nucleic acids, and other cofactors. It contributes to the energy balance by supplying ATP and NADPH to the cell and, therefore, it can confer “metabolic plasticity” by allowing a cell to adjust its nutrient status based on redox and epigenetic statuses
[33]. For these reasons, 1C metabolism is upregulated in cancer cells and is a major player in tumor proliferation (
Figure 9).
Figure 9. Regulation of one-carbon (1C) metabolism in cancer. Serine and glycine are transported into the cell or synthesized in the cell and serve as input molecules for 1C metabolism. Serine is synthesized from the glycolytic intermediate, 3-phosphoglycerate, via the de novo serine biosynthetic pathway (SSP) and glycine can be synthesized from serine via serine hydroxymethyltransferase 1 (SHMT1) in the cytoplasm or SHMT2 in the mitochondria. Upregulation of the SSP in cancer occurs via cMyc, Nrf2, and ATF4. Upregulation of the mitochondrial isoform SHMT2 occurs in cancer via hypoxia-inducible factor 1α (HIF-1α) and mutant KRAS-dependent pathways. Serine and glycine can both enter the early part of 1C metabolism, the folate cycle, although cancer cells tend to preferentially utilize serine over glycine. Upon entering the folate cycle, serine and glycine can donate 1C units to tetrahydrofolate (THF) to form 5,10-methylenetetrahydrofolate (me-THF). From there, one of three transformations can occur that lead to thymidylate synthesis, purine synthesis, or methionine synthesis via the coupling of the methionine synthase (MTR) reaction to conversion of homocysteine (hCYS) to form methionine (MET). The latter connects the folate cycle to the methionine cycle. The methionine cycle can be used to generate glutathione, proteins, and S-adenosylmethionine (SAM), all of which are important for cancer cell growth. ATP; adenosine tripihosphate, BHMT; betaine-homocysteine S-methyltransferase, DHFR; dihydrofolate reductase, F-THF; 10-formyltetrahydrofolate, GLDC; glycine dehydrogenase, HMT; histone methyl transferase, MAT; methionine adenosyltransferase, m-THF; 5-methyl-tetrahydrofolate, MTHFD 1/2/2L; methyltetrahydrofolate dehydrogenase 1/2/2L, MTHFR; methylenetetrahydrofolate reductase, NADPH; nicotinamide adenine dinucleotide phosphate, PHGDH; phosphoglycerate dehydrogenase, PSAT; phosphoserine aminotransferase; PSPH; phosphoserine phosphatase, SAH; S-adenosyl homocysteine, SAHH; SAH hydrolase. Figure created with
BioRender.com (accessed on 26 March 2021).
9. Other NEAAs
In addition to NEAAs that are heavily studied, i.e., serine, glycine, glutamate, and glutamine, the NEAAs alanine, aspartate, asparagine, arginine, cysteine, and proline are emerging as players in the tumor metabolic landscape [34].
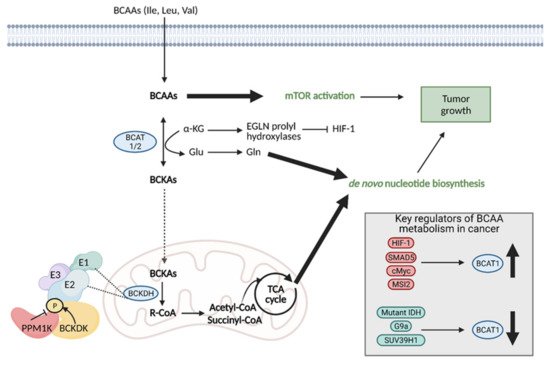
10. Branched Chain Amino Acid (BCAA) Metabolism
Branched chain amino acids (BCAAs) include the essential amino acids valine, leucine, and isoleucine and they play an important role in tumor cell growth and proliferation [35]. BCAAs function as nitrogen donors to produce nucleotides, can be incorporated into proteins, and can be broken down to produce important cell metabolites, such as glutamate, associating them with metabolic pathways critical for cancer progression [36]. Reprogrammed BCAA metabolism is involved in several types of cancer, including PDAC [37], glioblastoma [38], chronic myeloid leukemia (CML) [39], and endometrial cancer [40].
Regardless of the mechanism preferred, the enzymes involved in the first step of BCAA degradation are upregulated in cancer and they are reversible. This includes cytosolic branched-chain aminotransferase (BCAT1) and mitochondrial branched-chain aminotransferase (BCAT2), which convert BCAAS into branched-chain α-keto acids (BCKAs) by transferring the amino group to α-KG to generate glutamate or the reverse reaction [36]. Specifically, BCAT1 expression has been implicated in cancer growth, with suppression of BCAT1 limiting proliferation. BCAT1 expression is regulated by a number of molecules, including upregulation by HIF-1, SMAD5, c-Myc, and Musashi2 (MSI2), and downregulation by mutant IDH, and histone modifiers, G9a, and SUV39H1 (Figure 10) [35].
Figure 10. Reprogramming of branched chain amino acid (BCAA) metabolism in cancer. Branched chain amino acids (BCAAs), including isoleucine (Ile), leucine (Leu), and valine (Val), can be transported into the cell where they can directly activate mTOR signaling for tumor growth. They can also be converted to branched chain α-keto acids (BCKAs) via cytosolic branched chain amino acid transaminase 1 (BCAT1) or mitochondrial BCAT2 in a reversible reaction. BCAT1 overexpression results in increased BCAA catabolism, which is typical in cancer and is upregulated by several molecules (HIF-1, SMAD5, cMyc, MSI2), although some cancers favor the reverse reaction. The conversion of BCAAs to BCKAs generates glutamate, which can be used for de novo nucleotide biosynthesis. BCKAs can be further degraded in the mitochondria to acetyl CoA and succinyl CoA to power the TCA cycle and de novo nucleotide biosynthesis to support cancer proliferation. α-KG; alpha-ketoglutarate, BCKDH; branched-chain alpha-keto acid dehydrogenase complex, Glu; glutamate, Gln; glutamine, HIF-1; hypoxia-inducible factor 1, IDH; isocitrate dehydrogenase, MSI2; Musashi2, mTOR; mammalian target of rapamycin; PPM1K; Mg2+/Mn2+- dependent 1 K protein phosphatase, R-CoA; R-coenzyme A. Figure created with
BioRender.com (accessed on 26 March 2021).
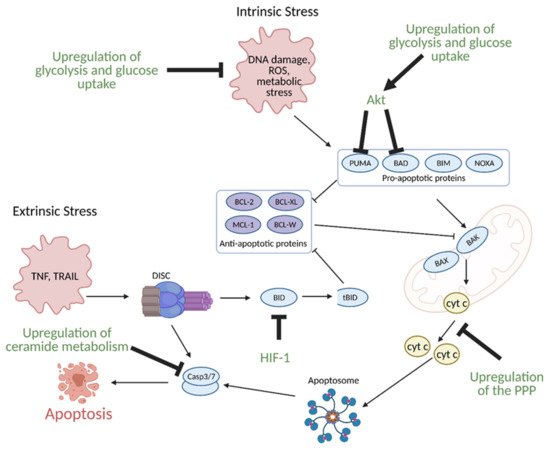
11. Regulation of Apoptosis by Metabolism
While healthy cells typically undergo programmed cell death or apoptosis during regular progression of the cell cycle, cancer cells evade apoptosis to enhance growth, proliferation, and survival under hypoxic conditions. Many of their apoptosis evasion mechanisms are linked to metabolic reprogramming
[41].
Glucose metabolism, specifically enhanced glycolysis, is one of the main hallmarks of metabolic reprogramming in cancer. Glucose metabolism is linked to the evasion of apoptosis in several ways. Many of the same signaling molecules involved in the upregulation of glycolysis are also involved in the suppression of apoptosis. For example, the hypoxic tumor microenvironment induces expression of aforementioned HIF-1, which in turn, leads to overexpression of glycolytic enzymes. Upregulation of glycolysis and glucose uptake are linked to resistance to apoptosis as increased glycolysis and glucose uptake prevents oxygen related damage to the cell, and thus, results in decreased apoptosis. HIF-1 also directly induces resistance to apoptosis via suppression of pro-apoptotic B cell lymphoma 2 (BCL-2) family protein BH3 interacting-domain death agonist (BID) (
Figure 11)
[42][43].
Figure 11. Regulation of apoptosis through metabolism in cancer. Apoptosis can be induced via intrinsic or extrinsic stress. Intrinsic stress results in the activation of pro-apoptotic proteins that then activate B-cell lymphoma 2 (Bcl-2)-associated X protein (BAX) and Bcl-2-homologous antagonist killer (BAK) in the mitochondria to facilitate cytochrome c (cyt c) release via mitochondrial outer membrane polarization. Cytochrome c release results in the formation of the apoptosome, which leads to the activation of caspase 3 (Casp3) and caspase 7 (Casp7). Casp3 and Casp7 then induce apoptosis. Extrinsic stress results in the formation of the Death Inducing Signaling Complex (DISC), which can directly activate apoptosis or trigger intrinsic apoptosis via BH3 interacting-domain death agonist (BID) cleavage to truncated BID (tBID). Cancer avoids apoptosis through multiple metabolism-mediated mechanisms, including reduction of reactive oxygen species (ROS) via the upregulation of glycolysis and glucose uptake, inhibition of pro-apoptotic proteins via growth factor signaling, inhibition of cyt c release via upregulation of the pentose phosphate pathway (PPP), inhibition of BID cleavage via hypoxia-inducible factor 1 (HIF-1), and inhibition of effector caspases via the upregulation of ceramide metabolism. Akt; protein kinase B, BAD; Bcl-2 associated death promoter, BIM; Bcl-2-like protein 11, BCL-W; Bcl-like protein 2, BCL-XL; Bcl-extra-large, MCL-1; myeloid-cell leukemia 1, NOXA; phorbol-12-myristate-13-acetate-induced protein 1, PUMA; p53 upregulated modulator of apoptosis. Figure created with
BioRender.com (accessed on 26 March 2021).
12. Role of Non-Coding RNAs in Cancer Cell Metabolism
Although the role of oncogenes, transcription factors, and other downstream signaling molecules has been widely established in mechanisms of cancer metabolic reprogramming, non-coding (nc) RNAs are emerging as important players. As ncRNAs were initially considered to lack biological function because they do not encode proteins, they play a role in cancer progression by regulating enzymes and pathways involved in the metabolic reprogramming of cancer cells. This regulation primarily occurs through glucose, glutamine, and lipid metabolism and involves two types of ncRNA, long-chain non-coding RNA (lncRNA) and microRNA (miRNA) [44].
13. Regulation of Cancer Growth via Tumor-Host Cell Metabolic Interactions
The reprogramming of metabolic pathways in cancer involves not only tumor cells themselves but also interactions between tumor cells and host cell populations
[45]. The tumor microenvironment is dense and includes fibroblasts, macrophages, mesenchymal stem cells, endothelial cells, and immune cells in addition to cancer cells
[46]. Heterocellular metabolic interactions between these populations in the tumor microenvironment work cohesively to support tumor growth and proliferation. With several different cell populations taking up residence, the tumor microenvironment faces many challenges and limiting factors for survival, including physical pressure, oxidative stress, nutrient deprivation, competition, and immune surveillance. To surmount these challenges and achieve tumor progression, tumor cells take advantage of the diverse microenvironment and engage in complex crosstalk with surrounding cells via nutrient sharing and metabolic symbiosis, competition, and the use of metabolites as signaling molecules
[45].
The sharing of molecules between different populations of cells in the tumor microenvironment allows for cancer cell growth by maximizing use of available nutrients for energy and macromolecule biosynthesis, providing alternative energy sources, and influencing immune cell populations.
Nutrient recycling between tumor cells and fibroblasts is also important for tumor growth. The main function of fibroblasts is to produce and secrete extracellular matrix, which increases the diversity of macromolecules surrounding the tumor cells
[45].
While many symbiotic tumor-host cell metabolic interactions contribute to tumor growth, competition between tumor and host cells for nutrients also significantly influences cancer progression
[47]. In order to carry out biosynthetic and bioenergetic activities, immune, tumor and stromal cells must all compete for nutrients. Immune cells are not adapted for competition, which leads to tumor growth via a loss of anti-tumor immune surveillance
[45].
14. Conclusions
Much progress has been made in the field of cancer metabolism since Warburg’s initial observations about 100 years ago. While cancer cells were previously thought to perform only aerobic glycolysis, it is now evident their growth and proliferation is dependent on the reprogramming of a large number of pathways, such as the TCA cycle, the PPP, lipogenesis, 1C metabolism, and BCAA metabolism, as well as the utilization of alternate substrates, conferring them with metabolic plasticity. Altered flux through metabolic pathways is coordinated by many different genes and regulatory molecules that work together to support enhance growth and proliferation. Crosstalk between many of these pathways is evident, including that between glycolysis and the PPP and the regulation of apoptosis through metabolism. Understanding the complex nature of altered metabolic flux in cancer cells and its context dependent nature is crucial to the development and application of new therapies.
 +1 credit
+1 credit