 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ivan Verduci | + 1973 word(s) | 1973 | 2021-07-15 09:49:27 | | | |
| 2 | Lindsay Dong | Meta information modification | 1973 | 2021-07-27 04:27:21 | | | | |
| 3 | Conner Chen | Meta information modification | 1973 | 2021-10-11 08:46:45 | | |
Video Upload Options
Identification of potential pathological biomarkers has proved to be essential for understanding complex and fatal diseases, such as cancer and neurodegenerative diseases. Ion channels are involved in the maintenance of cellular homeostasis. Moreover, loss of function and aberrant expression of ion channels and transporters have been linked to various cancers, and to neurodegeneration. The Chloride Intracellular Channel 1 (CLIC1), CLIC1 is a metamorphic protein belonging to a partially unexplored protein superfamily, the CLICs. In homeostatic conditions, CLIC1 protein is expressed in cells as a cytosolic monomer. In pathological states, CLIC1 is specifically expressed as transmembrane chloride channel. In the following review, we trace the involvement of CLIC1 protein functions in physiological and in pathological conditions and assess its functionally active isoform as a potential target for future therapeutic strategies.
1. CLIC Proteins, a Focus on CLIC1
Chloride channels exert different functions in every stage. They are involved in ion homeostasis, fluid transportation, regulation of cell volume, cytoskeletal rearrangement, and cellular motility. An example is the interplay between swelling-activate K+ and Cl- channels during volume regulation due to cellular hypertonicity.
Chloride Intracellular Channel proteins (CLIC) are a class of proteins both soluble and integral membrane forms. Littler et al. showed CLIC proteins to be highly conserved in chordates with the presence of six vertebrate paralogues [1]. CLIC proteins diverge from the canonical ion channel structure. Using phylogen and structural techniques they found CLIC proteins exert enzymatic functions. CLIC proteins show 240 aminoacidic sequence belonging to the glutathione S-transferase (GST) fold superfamily. This suggests a well-conserved intracellular activity playing a role in redox balance. In particular, the enzymatic active site was shown to be the Cys24, distinct from canonical GSTs which has been shown to exhibit a thiol group [2]. As a variety of metamorphic proteins, CLIC proteins undergo important changes in structure switching from the globular hydrophilic form to the transmembrane hydrophobic structure. It was found that the membrane insertion is elicited by oxidizing conditions and pH changes [2][3][4][5]. Oxidation-dependent insertion is likely to be mediated by the active residue Cys24 as the C24A mutation alters the redox sensitivity of the channel and—for CLIC1—its electrophysiological characteristics. CLICs channel activity at physiological pH has been shown to be minimal while the presence of membrane-expressed rises with a change in pH [4].
CLIC proteins were found to also be involved in endothelium architecture. Tavasoli and colleagues have demonstrated that both CLIC1 and CLIC5A activate ezrin, radixin, and moesin proteins for the formation of the lumen of vessels. As a result, the dual CLIC4/CLIC5-deficient mice developed spontaneous proteinuria, glomerular cell proliferation, and matrix deposition [6]. Among CLIC proteins, despite CLIC1 is considered a chloride channel, its RNA transcripts lack the specific sequences for membrane insertion. Thus, it does not follow secretary pathways typical of canonical ion channels through endoplasmic reticulum and Golgi apparatus [7]. For this reason, to demonstrate that CLIC1 protein is a channel, rather than a modulatory element, recombinant protein inserted in artificial lipid bilayers has been used. These experiments evidenced a conductance in internal–external 140 mM KCl of 30 pS with a variety of substates suggesting that the integral membrane form of CLIC1 would comprise four single monomers [2][3][5][7][8].
2. CLIC1 in Non-Pathological States
More recently, relatively abundant levels of CLIC proteins have been found in human bronchial epithelial cells primary cultures. In particular, it has been shown CLIC1 to be able to modulate cAMP-induced chloride currents [9]. The authors show that one of the well-known CFTR antagonists on the market, PPQ-102 abolished the currents elicited by isoproterenol or forskolin. Despite the fact that this effect is attributed, usually, to the activation of CFTR, RNA Seq datasets show the absence of this transcript in the analyzed cells. As a matter of fact, knockdown of CLIC1 inhibits significantly cAMP-induced chloride currents, suggesting a direct contribution of CLIC1 to this chloride conductance. The authors concluded that manipulating CLIC1 properties may enhance Cl- conductance, thus proposing the possibility to potentiate CLIC1 functional activity as a possible therapeutic strategy in conditions of impaired chloride homeostasis as cystic fibrosis [9].
3. Activation of Transitory Allostasis through CLIC1 Function
In tissues, CLIC1 is found mainly as a cytoplasmic protein able to shuttle to plasma membrane following a remodeling elicited by perturbation of cellular homeostasis. The two major players are oxidative stress and pH alterations. Overproduction of reactive oxygen species (ROS), modifies the structure of the protein through the formation of a disulphide bridge between Cys24 and Cys59, considered the essential oxidoreductase residues, which promotes membrane docking [3][10][11]. Transient oxidative stress is proper of physiological functions as cell cycle progression [12] and innate immune responses [13].
The transient translocation of CLIC1 in plasma membrane of macrophages promotes phagosome acidification from 4.4 to 3.4. Clic1−/− macrophages display impaired phagosome proteolysis and altered ability to kill microorganisms and to expose antigens to T cells [14]. Moreover, it has been demonstrated that tmCLIC1 activity contributes to the formation of inflammasome modulating NLRP3 complex [15][13], a multiproteic complex crucial for the immune system, which regulates the activation of Caspase 1 and release of cytokines as Interleukine 6 (IL-6) and Interleukine 8 (IL-8), promoting pyroptosis (inflammatory programmed cell death).
4. CLIC1 during Chronic Allostasis
4.1. CLIC1 in Solid Tumors
Lu and colleagues have postulated that CLIC1 acts as an oncogene in pancreatic cancer. Here, patients with CLIC1-positive tumors have demonstrated worse overall survival compared to those with CLIC1-negative tumors [16]. CLIC1 expression is correlated to a poor prognosis not only in pancreatic cancer, but also in tumors as lung cancer [17], ovarian cancer, where CLIC1 upregulation was correlated to chemotherapy resistance [18], gallbladder, and gastric cancers [19], where it was found to promote cells proliferation via MAPK/AKT regulation [20] and facilitates the formation of tumor-associated fibroblasts [21]. In addition, CLIC1 protein upregulation correlates with the level of aggressiveness and metastatic potential of colorectal cancer cells [22], where it was demonstrated to regulate cell volume and ROS level. One of the aspects of cancer is its heterogeneity, it is composed of differentiated cells, representing the major component of the tumor mass, and cancer stem cells (CSCs) [23]. Despite cancer stem cells constitute a small percentage of tumor cells (0.05–1%), they are the prime sources of tumor recurrence and metastasis as they confer resistance to chemo and radiotherapies [24]. Cancer stem cells are able to take advantage of the aberrant redox system. In particular, it was demonstrated that oxidative stress and gene-environment interactions support the development of a huge variety of solid tumors, as glioblastoma, breast, prostate, pancreatic, and colon cancer [25].
4.2. CLIC1 in Neurodegenerative Processes
The first evidence about tmCLIC1 involvement in neurodegenerative processes dates to 2004 [26]. In the paper, the authors show an increase in protein expression, and enhanced functional activity in response to Aβ peptide incubation in microglial cells. In addition, the Aβ-induced release of Nitric Oxide (NO), as well as TNF-α is prevented by impairing tmCLIC1 function by its pharmacological inhibition and by RNA interference, supporting an active role of the protein during the neurodegenerative processes. A possible mechanism by which tmCLIC1 would support the neurodegenerative process was suggested in a following work [27]. In particular, its ion channel activity would sustain the Aβ-induced ROS production by microglial cells, acting as a charge compensator, which balances the electrogenic activity of the NADPH Oxidase (NOX2). The correlation between tmCLIC1 functional activity and neurodegeneration was further confirmed by the observation of high protein levels in section from 3xTg-AD mouse brain, characterized by a progressive deposition of Aβ during their life. The authors concluded that tmCLIC1 inhibition could represent a promising anti-inflammatory target for Alzheimer’s Disease therapy. This was further strengthened by the work of Paradisi et al. [28] where the authors have shown that the inhibition of tmCLIC1 activity by pharmacological blockers and the suppression by RNA interference does not alter the activation of resident microglia. In particular, they show that affecting tmCLIC1 ion channel produces a reduction in some toxic aspects of activated microglia (i.e., NO production) while leaving unaltered its phagocytic ability. Moreover, in a following work it was shown that by co-culturing neurons and microglia in a two-chamber transwell, IAA94 treatment was able to prevent the neurotoxicity induced by the treatment with Aβ1-42 peptide [29].
5. Conclusions
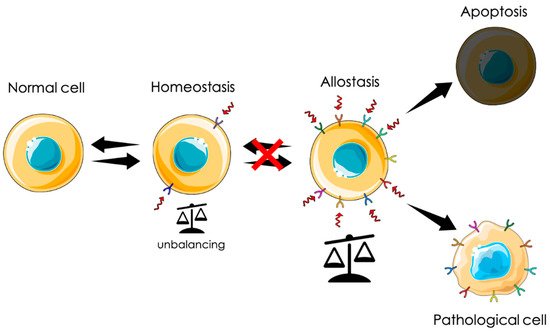
From a physiological point of view, tumorigenesis can be defined as an adaptation. Uncontrolled proliferation can be seen as an extreme survival mechanism in response to a chronic stress state generated by a hostile environment. In this scenario, among other proteins, CLIC1 is instrumental for this transformation. In particular, as a metamorphic protein, its translocation to the plasma membrane could be a first line response to diverse stimuli. Transmembrane insertion and the consequent increased ionic permeability can represent a fast reaction to counteract cytoplasmic unbalance conditions. As mentioned before, cytoplasmic CLIC1 protein binds GSH. In adverse conditions, the rearrangement of the protein structure releases GSH, contributing to buffer cytoplasmic oxidation. At the same time, CLIC1 association with an ionic channel guarantees the possibility to disperse any excess of charges that could accumulate during a persistent condition of oxidative stress. In the central nervous system, the presence of high concentration of β-amyloid generates a state of oxidative stress that triggers microglia activation with the intent of phagocyte oligomers and fibrils. The release of a huge amount of amyloid causes microglia hyperactivation that becomes harmful for neurons and glial cells, generating the process of neurodegeneration. In solid tumors, the process may be different. Chronic conditions of oxidative stress due to environmental causes like pollution, smoke, wrong or excessive alimentation, or hormonal dysregulation could induce cell “adaptation” giving rise to a neoplastic process. Both these conditions show a direct correlation between the presence of membrane CLIC1 and the state of microglia activation or tumors aggressiveness. The initial mechanism of neurodegeneration and tumorigenesis in sporadic cases is still obscure. However, CLIC1 protein functional expression as a membrane protein could be symptomatic of hyperactivated states as that of neurodegeneration or the neoplastic transformation. In this perspective, tmCLIC1 could represent a promising element as a diagnostic tool, as well as a therapeutic target.
References
- Littler, D.R.; Harrop, S.J.; Goodchild, S.; Phang, J.M.; Mynott, A.V.; Jiang, L.; Valenzuela, S.; Mazzanti, M.; Brown, L.; Breit, S.N.; et al. The enigma of the CLIC proteins: Ion channels, redox proteins, enzymes, scaffolding proteins? FEBS Lett. 2010, 584, 2093–2101.
- Harrop, S.J.; DeMaere, M.; Fairlie, W.; Reztsova, T.; Valenzuela, S.; Mazzanti, M.; Tonini, R.; Qiu, M.R.; Jankova, L.; Warton, K.; et al. Crystal Structure of a Soluble Form of the Intracellular Chloride Ion Channel CLIC1 (NCC27) at 1.4-Å Resolution. J. Biol. Chem. 2001, 276, 44993–45000.
- Littler, D.R.; Harrop, S.J.; Fairlie, W.; Brown, L.; Pankhurst, G.J.; Pankhurst, S.; DeMaere, M.; Campbell, T.; Bauskin, A.R.; Tonini, R.; et al. The Intracellular Chloride Ion Channel Protein CLIC1 Undergoes a Redox-controlled Structural Transition. J. Biol. Chem. 2004, 279, 9298–9305.
- Tulk, B.M.; Kapadia, S.; Edwards, J.C. CLIC1 inserts from the aqueous phase into phospholipid membranes, where it functions as an anion channel. Am. J. Physiol. Cell Physiol. 2002, 282, C1103–C1112.
- Warton, K.; Tonini, R.; Fairlie, W.D.; Matthews, J.M.; Valenzuela, S.M.; Qiu, M.R.; Wu, W.M.; Pankhurst, S.; Bauskin, A.R.; Harrop, S.J.; et al. Recombinant CLIC1 (NCC27) assembles in lipid bilayers via a pH-dependent two-state process to form chloride ion channels with identical characteristics to those observed in Chinese hamster ovary cells expressing CLIC1. J. Biol. Chem. 2002, 277, 26003–26011.
- Tavasoli, M.; Al-Momany, A.; Wang, X.; Li, L.; Edwards, J.C.; Ballermann, B.J. Both CLIC4 and CLIC5A activate ERM proteins in glomerular endothelium. Am. J. Physiol. Renal Physiol. 2016, 311, F945–F957.
- Valenzuela, S.; Martin, D.K.; Por, S.B.; Robbins, J.M.; Warton, K.; Bootcov, M.; Schofield, P.; Campbell, T.J.; Breit, S.N. Molecular Cloning and Expression of a Chloride Ion Channel of Cell Nuclei. J. Biol. Chem. 1997, 272, 12575–12582.
- Singh, H.; Ashley, R.H. Redox Regulation of CLIC1 by Cysteine Residues Associated with the Putative Channel Pore. Biophys. J. 2006, 90, 1628–1638.
- Liu, B.; Billington, C.K.; Henry, A.P.; Bhaker, S.K.; Kheirallah, A.K.; Swan, C.; Hall, I. Chloride intracellular channel 1 (CLIC1) contributes to modulation of cyclic AMP-activated whole-cell chloride currents in human bronchial epithelial cells. Physiol. Rep. 2018, 6.
- Al Khamici, H.; Hossain, K.R.; Cornell, B.A.; Valenzuela, S.M. Investigating Sterol and Redox Regulation of the Ion Channel Activity of CLIC1 Using Tethered Bilayer Membranes. Membranes 2016, 6, 51.
- Al Khamici, H.; Brown, L.J.; Hossain, K.R.; Hudson, A.L.; Sinclair-Burton, A.A.; Ng, J.P.M.; Daniel, E.L.; Hare, J.E.; Cornell, B.A.; Curmi, P.M.G.; et al. Members of the Chloride Intracellular Ion Channel Protein Family Demonstrate Glutaredoxin-Like Enzymatic Activity. PLoS ONE 2015, 10, e115699.
- Patterson, J.C.; Joughin, B.A.; Van De Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst. 2019, 8, 163–167.e2.
- Tang, J.; Hu, P.; Li, Y.; Win-Shwe, T.-T.; Li, C. Ion Imbalance Is Involved in the Mechanisms of Liver Oxidative Damage in Rats Exposed to Glyphosate. Front. Physiol. 2017, 8, 1083.
- Jiang, L.; Salao, K.; Li, H.; Rybicka, J.M.; Yates, R.M.; Luo, X.W.; Shi, X.X.; Kuffner, T.; Tsai, V.W.-W.; Husaini, Y.; et al. Intracellular chloride channel protein CLIC1 regulates macrophage functions via modulation of phagosomal acidification. J. Cell Sci. 2012, 125, 5479–5488.
- Domingo-Fernández, R.; Coll, R.C.; Kearney, J.; Breit, S.; O’Neill, L.A. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1beta transcription and activate the NLRP3 inflammasome. J. Biol. Chem. 2017, 292, 12077–12087.
- Lu, J.; Dong, Q.; Zhang, B.; Wang, X.; Ye, B.; Zhang, F.; Song, X.; Gao, G.; Mu, J.; Wang, Z.; et al. Chloride intracellular channel 1 (CLIC1) is activated and functions as an oncogene in pancreatic cancer. Med. Oncol. 2015, 32, 171.
- Lee, J.R.; Lee, J.Y.; Kim, H.J.; Hahn, M.J.; Kang, J.S.; Cho, H. The inhibition of chloride intracellular channel 1 enhances Ca(2+) and reactive oxygen species signaling in A549 human lung cancer cells. Exp. Mol. Med. 2019, 51, 1–11.
- Yu, W.; Cui, R.; Qu, H.; Liu, C.; Deng, H.; Zhang, Z. Expression and prognostic value of CLIC1 in epithelial ovarian cancer. Exp. Ther. Med. 2018, 15, 4943–4949.
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772.
- Li, B.-P.; Mao, Y.-T.; Wang, Z.; Chen, Y.-Y.; Wang, Y.; Zhai, C.-Y.; Shi, B.; Liu, S.-Y.; Liu, J.-L.; Chen, J.-Q. CLIC1 Promotes the Progression of Gastric Cancer by Regulating the MAPK/AKT Pathways. Cell. Physiol. Biochem. 2018, 46, 907–924.
- Meng, M.; Wang, H.B.; Song, S.Z.; Sun, R.; Lin, Y.; Lin, S. CLIC1 facilitates cancer associated fibroblast activation and gastric cancer progression via integrins/NF-kappaB pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 320, G836.
- Wang, P.; Zhang, C.; Yu, P.; Tang, B.; Liu, T.; Cui, H.; Xu, J. Regulation of colon cancer cell migration and invasion by CLIC1-mediated RVD. Mol. Cell. Biochem. 2012, 365, 313–321.
- Setti, M.; Savalli, N.; Osti, D.; Richichi, C.; Angelini, M.; Brescia, P.; Fornasari, L.; Carro, M.S.; Mazzanti, M.; Pelicci, G. Functional Role of CLIC1 Ion Channel in Glioblastoma-Derived Stem/Progenitor Cells. J. Natl. Cancer Inst. 2013, 105, 1644–1655.
- Ding, Q.; Li, M.; Wu, X.; Zhang, L.; Wu, W.; Ding, Q.; Weng, H.; Wang, X.; Liu, Y. CLIC1 overexpression is associated with poor prognosis in gallbladder cancer. Tumour Biol. 2015, 36, 193–198.
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxid. Med. Cell Longev. 2010, 3, 23–34.
- Novarino, G.; Fabrizi, C.; Tonini, R.; Denti, M.A.; Malchiodi-Albedi, F.; Lauro, G.; Sacchetti, B.; Paradisi, S.; Ferroni, A.; Curmi, P.M.; et al. Involvement of the intracellular ion channel CLIC1 in microglia-mediated beta-amyloid-induced neurotoxicity. J. Neurosci. 2004, 24, 5322–5330.
- Milton, R.H.; Abeti, R.; Averaimo, S.; DeBiasi, S.; Vitellaro, L.; Jiang, L.; Curmi, P.M.G.; Breit, S.N.; Duchen, M.R.; Mazzanti, M. CLIC1 function is required for beta-amyloid-induced generation of reactive oxygen species by microglia. J. Neurosci. 2008, 28, 11488–11499.
- Paradisi, S.; Matteucci, A.; Fabrizi, C.; Denti, M.A.; Abeti, R.; Breit, S.N.; Malchiodi-Albedi, F.; Mazzanti, M. Blockade of chloride intracellular ion channel 1 stimulates Aβ phagocytosis. J. Neurosci. Res. 2008, 86, 2488–2498.
- Skaper, S.D.; Facci, L.; Giusti, P. Intracellular ion channel CLIC1: Involvement in microglia-mediated beta-amyloid peptide(1-42) neurotoxicity. Neurochem. Res. 2013, 38, 1801–1808.


