 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natália Cruz-Martins | + 5138 word(s) | 5138 | 2021-07-22 08:30:27 | | | |
| 2 | Vivi Li | Meta information modification | 5138 | 2021-07-22 11:44:01 | | |
Video Upload Options
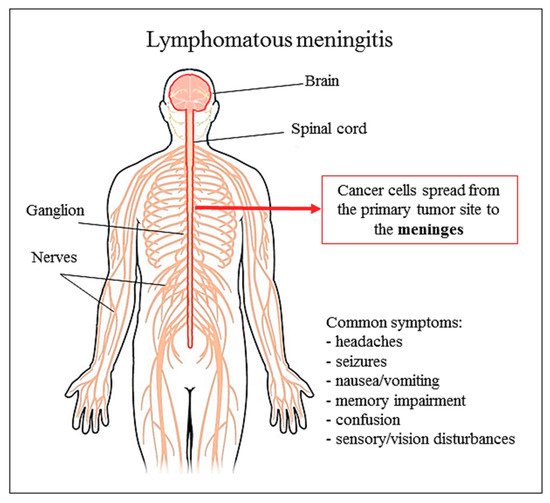
Cancer is the second leading cause of death worldwide. The main modality to fight against cancer is surgery, radiotherapy, and chemotherapy, and more recently targeted therapy, gene therapy and immunotherapy, which play important roles in treating cancer patients. In the last decades, chemotherapy has been well developed. Nonetheless, administration of the drug is not always successful, as limited drug dosage can reach the tumor cells.. In this context, the possibility to use an encapsulated anti-cancer drug may potentially solve the problem. Liposomal cytarabine is a formulation with pronounced effectiveness in lymphomatous meningitis and reduced cardiotoxicity if compared to liposomal anthracyclines. Thus, the future liposomal cytarabine use could be extended to other diseases given its reduction in cytotoxic side effects compared to the free formulation.
1. Introduction
| Phase | Treatment | Disease | Enrollment | Identifier |
|---|---|---|---|---|
| Phase 2 | DepoCyt, methotrexate | Leptomeningeal metastasis of breast cancer | 3 | NCT00992602 |
| Phase 2 | DepoCyt | Lymphomatous or leukemic meningitis | 4 | NCT00523939 |
| Not Applicable | DepoCyt, sorafenib | Neoplastic meningitis | 2 | NCT00964743 |
| Phase 3 | DepoCyt | Leptomeningeal metastasis of breast cancer | 74 | NCT01645839 |
| Not Applicable | Vyxenos, liposomal cytarabine, and daunorubicin | Untreated myelodysplastic syndrome, acute myeloid leukemia, acute biphenotypic leukemia, myelodysplastic syndrome | 48 | NCT01804101 |
| Phase 3 | Vyxenos, 7+3 (liposomal cytarabine and daunorubicin) | High risk of acute myeloid leukemia | 309 | NCT01696084 |
2. Chemistry of Liposomes: From Models to New Applications
3. Liposomal Cytarabine
3.1. Preclinical Data and Research
3.2. Clinical Use
| Study Type | Treatment | Disease | Results | Ref. |
|---|---|---|---|---|
| Phase I | Intraventricular | Leptomeningeal metastasis | Well-tolerated toxicity, duration of response with a median of over 11 weeks | [65] |
| Phase I | Intrathecal | Neoplastic meningitis | The therapeutic intra-lumbar concentration of free Ara-C was maintained for up to 14 days | [53] |
| Phase I | Intra-lumbar | Leptomeningeal metastasis | Extended free Ara-C concentrations | [66] |
| Phase II | Intrathecal | Leptomeningeal metastasis | Well-tolerated toxicity, systemic high-dose methotrexate + liposomal cytarabine | [67] |
| Randomized controlled trial | Intrathecal | Neoplastic meningitis | Increased time to neurological progression. Median survival was 105 days with DepoCyt and 78 days with methotrexate | [68] |
| Open-label study | Intraventricular or lumbar puncture | Leukemia, lymphoma, or solid tumors as part of a phase III study | Extended exposure compared with standard Ara-C | [69] |
| Case-report | Intrathecal | Secondary diffuse leptomeningeal gliomatosis | Improvement of the clinical status | [70] |
| Retrospective case series | Intraventricular | Leptomeningeal metastasis | Well tolerate toxicity, in general | [52] |
| Study Type | No. of Patients | Results | Reference |
|---|---|---|---|
| Phase I | 9 | Duration of response: 2–14 weeks, median 11 | [65] |
| Phase I | 12 | Therapeutic intralumbar concentration of free cytarabine maintained for 14 days | [53] |
| Phase I | 8 | Lumbar and intraventricular max concentration of free cytarabine: 226 and 6.06 mg/L; half-life, 277 and 130 h, respectively | [66] |
| Phase I | 18 children (3–21 years) | Prolonged disease stabilization or response: 14 patients Maximum-tolerated dose: 35 mg |
[73] |
| Randomized controlled trial | 31 treated with D, 30 with M | Median survival: 105 days (D), 78 days (M) Median time to neurological progression: 58 (D) vs 30 (M) days Neoplastic meningitis-specific survival: 343 (D) versus 98 (M) days Adverse events: comparable D vs. M |
[68] |
| Open-label study | 8 | Concentration of free and encapsulated cytarabine in the ventricular and lumbar CSF: 0.01 to 1500 µg/mL | [69] |
| Case-report | 1 | Duration of response with D: 6 months | [74] |
| Retrospective case series | 120 | D well tolerated, but 12.5% had serious treatment-related neurological complications | [52] |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA 2018, 68, 7–30.
- Salehi, B.; Zucca, P.; Sharifi-Rad, M.; Pezzani, R.; Rajabi, S.; Setzer, W.N.; Varoni, E.M.; Iriti, M.; Kobarfard, F.; Sharifi-Rad, J. Phytotherapeutics in cancer invasion and metastasis. Phytother. Res. 2018.
- Barabadi, H.; Alizadeh, A.; Ovais, M.; Ahmadi, A.; Shinwari, Z.; Muthupandian, S. The efficacy of green nanoparticles against cancerous and normal cell lines: A systematic review and meta-analysis. IET Nanobiotechnol. 2017, 12, 377–391.
- Barabadi, H.; Ovais, M.; Shinwari, Z.K.; Saravanan, M. Anti-cancer green bionanomaterials: Present status and future prospects. Green Chem. Lett. Rev. 2017, 10, 285–314.
- Mishra, A.P.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. Programmed Cell Death, from a Cancer Perspective: An Overview. Mol. Diagn. Ther. 2018, 22, 281–295.
- Yang, J.; Su, H.; Sun, W.; Cai, J.; Liu, S.; Chai, Y.; Zhang, C. Dual Chemodrug-Loaded Single-Walled Carbon Nanohorns for Multimodal Imaging-Guided Chemo-Photothermal Therapy of Tumors and Lung Metastases. Theranostics 2018, 8, 1966.
- Leriche, G.; Cifelli, J.L.; Sibucao, K.C.; Patterson, J.P.; Koyanagi, T.; Gianneschi, N.C.; Yang, J. Characterization of drug encapsulation and retention in archaea-inspired tetraether liposomes. Organ. Biomol. Chem. 2017, 15, 2157–2162.
- Siontorou, C.G.; Nikoleli, G.P.; Nikolelis, D.P.; Karapetis, S.K. Artificial Lipid Membranes: Past, Present, and Future. Membranes 2017, 7, 38.
- Weissig, V. Liposomes Came First: The Early History of Liposomology. Methods Mol. Biol. 2017, 1522, 1–15.
- Riaz, M.K.; Riaz, M.A.; Zhang, X.; Lin, C.; Wong, K.H.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Surface Functionalization and Targeting Strategies of Liposomes in Solid Tumor Therapy: A Review. Int. J. Mol. Sci. 2018, 19, 195.
- Bally, M.B.; Mayer, L.D.; Loughrey, H.; Redelmeier, T.; Madden, T.D.; Wong, K.; Harrigan, P.R.; Hope, M.J.; Cullis, P.R. Dopamine accumulation in large unilamellar vesicle systems induced by transmembrane ion gradients. Chem. Phys. Lipids 1988, 47, 97–107.
- Fatima, M.T.; Islam, Z.; Ahmad, E.; Barreto, G.E.; Md Ashraf, G. Ionic gradient liposomes: Recent advances in the stable entrapment and prolonged released of local anesthetics and anticancer drugs. Biomed. Pharmacother. 2018, 107, 34–43.
- Ma, P.; Dong, X.; Swadley, C.L.; Gupte, A.; Leggas, M.; Ledebur, H.C.; Mumper, R.J. Development of idarubicin and doxorubicin solid lipid nanoparticles to overcome Pgp-mediated multiple drug resistance in leukemia. J. Biomed. Nanotechnol. 2009, 5, 151–161.
- Gubernator, J. Active methods of drug loading into liposomes: recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin. Drug Deliv. 2011, 8, 565–580.
- Abraham, S.A.; McKenzie, C.; Masin, D.; Ng, R.; Harasym, T.O.; Mayer, L.D.; Bally, M.B. In vitro and in vivo characterization of doxorubicin and vincristine coencapsulated within liposomes through use of transition metal ion complexation and pH gradient loading. Clin. Cancer Res. 2004, 10, 728–738.
- Vabbilisetty, P.; Sun, X.-L. Liposome surface functionalization based on different anchoring lipids via Staudinger ligation. Org. Biomol. Cem. 2014, 12, 1237–1244.
- Nobs, L.; Buchegger, F.; Gurny, R.; Allémann, E. Current methods for attaching targeting ligands to liposomes and nanoparticles. J. Pharm. Sci. 2004, 93, 1980–1992.
- Richter, R.P.; Bérat, R.; Brisson, A.R. Formation of solid-supported lipid bilayers: an integrated view. Langmuir 2006, 22, 3497–3505.
- Troutier, A.-L.; Ladavière, C. An overview of lipid membrane supported by colloidal particles. Adv. Colloid Interf. Sci. 2007, 133, 1–21.
- Mornet, S.; Lambert, O.; Duguet, E.; Brisson, A. The formation of supported lipid bilayers on silica nanoparticles revealed by cryoelectron microscopy. Nano Lett. 2005, 5, 281–285.
- Peetla, C.; Stine, A.; Labhasetwar, V. Biophysical interactions with model lipid membranes: applications in drug discovery and drug delivery. Mol. Pharm. 2009, 6, 1264–1276.
- Michot, J.-M.; Seral, C.; Van Bambeke, F.; Mingeot-Leclercq, M.-P.; Tulkens, P.M. Influence of efflux transporters on the accumulation and efflux of four quinolones (ciprofloxacin, levofloxacin, garenoxacin, and moxifloxacin) in J774 macrophages. Antimicrob. Agents Chemother. 2005, 49, 2429–2437.
- Bensikaddour, H.; Snoussi, K.; Lins, L.; Van Bambeke, F.; Tulkens, P.M.; Brasseur, R.; Goormaghtigh, E.; Mingeot-Leclercq, M.-P. Interactions of ciprofloxacin with DPPC and DPPG: fluorescence anisotropy, ATR-FTIR and 31 P NMR spectroscopies and conformational analysis. Biochim. Biophys. Acta 2008, 1778, 2535–2543.
- Fa, N.; Lins, L.; Courtoy, P.J.; Dufrêne, Y.; Van Der Smissen, P.; Brasseur, R.; Tyteca, D.; Mingeot-Leclercq, M.-P. Decrease of elastic moduli of DOPC bilayers induced by a macrolide antibiotic, azithromycin. Biochim. Biophys. Acta 2007, 1768, 1830–1838.
- Klopman, G.; Zhu, H. Recent methodologies for the estimation of n-octanol/water partition coefficients and their use in the prediction of membrane transport properties of drugs. Mini Rev. Med. Chem. 2005, 5, 127–133.
- Rodrigues, C.; Gameiro, P.; Reis, S.; Lima, J.; de Castro, B. Derivative spectrophotometry as a tool for the determination of drug partition coefficients in water/dimyristoyl-L-α-phosphatidylglycerol (DMPG) liposomes. Biophys. Chem. 2001, 94, 97–106.
- Baciu, M.; Sebai, S.C.; Ces, O.; Mulet, X.; Clarke, J.A.; Shearman, G.C.; Law, R.V.; Templer, R.H.; Plisson, C.; Parker, C.A. Degradative transport of cationic amphiphilic drugs across phospholipid bilayers. Philos. Trans. Roy. Soc. Lond. A 2006, 364, 2597–2614.
- Pavinatto, F.J.; Caseli, L.; Pavinatto, A.; dos Santos, D.S.; Nobre, T.M.; Zaniquelli, M.E.; Silva, H.S.; Miranda, P.B.; de Oliveira, O.N. Probing chitosan and phospholipid interactions using Langmuir and Langmuir− Blodgett films as cell membrane models. Langmuir 2007, 23, 7666–7671.
- Yusupov, M.; Van der Paal, J.; Neyts, E.; Bogaerts, A. Synergistic effect of electric field and lipid oxidation on the permeability of cell membranes. Biochim. Biophys. Acta 2017, 1861, 839–847.
- Phillips, M.A.; Gran, M.L.; Peppas, N.A. Targeted nanodelivery of drugs and diagnostics. Nano Today 2010, 5, 143–159.
- Huynh, R.; Chaubet, F.; Jozefonvicz, J. Anticoagulant properties of dextranmethylcarboxylate benzylamide sulfate (DMCBSu); a new generation of bioactive functionalized dextran. Carbohydr. Res. 2001, 332, 75–83.
- Barrera, C.; Herrera, A.P.; Rinaldi, C. Colloidal dispersions of monodisperse magnetite nanoparticles modified with poly (ethylene glycol). J. Colloid Interface Sci. 2009, 329, 107–113.
- Gopalakrishnan, G.; Danelon, C.; Izewska, P.; Prummer, M.; Bolinger, P.Y.; Geissbühler, I.; Demurtas, D.; Dubochet, J.; Vogel, H. Multifunctional lipid/quantum dot hybrid nanocontainers for controlled targeting of live cells. Angew. Chem. 2006, 45, 5478–5483.
- Matsuno, R.; Ishihara, K. Integrated functional nanocolloids covered with artificial cell membranes for biomedical applications. Nano Today 2011, 6, 61–74.
- Thanh, N.T.; Green, L.A. Functionalisation of nanoparticles for biomedical applications. Nano Today 2010, 5, 213–230.
- Dubertret, B.; Skourides, P.; Norris, D.J.; Noireaux, V.; Brivanlou, A.H.; Libchaber, A. In vivo imaging of quantum dots encapsulated in phospholipid micelles. Science 2002, 298, 1759–1762.
- Hyodo, K.; Yamamoto, E.; Suzuki, T.; Kikuchi, H.; Asano, M.; Ishihara, H. Development of liposomal anticancer drugs. Biol. Pharm. Bull. 2013, 36, 703–707.
- Hamada, A.; Kawaguchi, T.; Nakano, M. Clinical pharmacokinetics of cytarabine formulations. Clin. Pharm. 2002, 41, 705–718.
- Evans, J.S.; Musser, E.A.; Mengel, G.D.; Forsblad, K.R.; Hunter, J.H. Antitumor activity of 1-beta-D-arainofuranosylcytosine hydrochloride. Proc. Soc. Exp. Biol. Med. 1961, 106, 350–353.
- Talley, R.W.; Vaitkevicius, V.K. Megaloblastosis produced by a cytosine antagonist, 1-beta-D-arabinofuranosylcytosine. Blood 1963, 21, 352–362.
- Newman, D.J.; Cragg, G.M.; Battershill, C.N. Therapeutic Agents from the Sea: Biodiversity, Chemo-Evolutionary Insight and Advances To the End of Darwin’s 200th Year; National Library of Medicine: Bethesda, MD, USA, 2009.
- Tyner, J.W.; Tardi, P.; Mayer, L.; Fletcher, L.B.; Spurgeon, S.; Kovacsovics, T.; Loriaux, M.M. Evaluation of CPX-351 (cytarabine: Daunorubicin) liposome injection anti-Leukemic activity against primary patient leukemia cells. Am. Soc. Hematol. 2010.
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38.
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Rel. 2012, 161, 175–187.
- Slingerland, M.; Guchelaar, H.-J.; Gelderblom, H. Liposomal drug formulations in cancer therapy: 15 years along the road. Drug Discov. Today 2012, 17, 160–166.
- Juzenas, P.; Chen, W.; Sun, Y.-P.; Coelho, M.A.N.; Generalov, R.; Generalova, N.; Christensen, I.L. Quantum dots and nanoparticles for photodynamic and radiation therapies of cancer. Adv. Drug Deliv. Rev. 2008, 60, 1600–1614.
- Bhojwani, D.; Pui, C.-h. Intrathecal liposomal cytarabine: More friend than foe? Leuk. Lymphoma 2008, 49, 1427–1430.
- Kratz, F.; Senter, P.; Steinhagen, H. Drug Delivery in Oncology: From Basic Research to Cancer Therapy; John Wiley & Sons: Hoboken, NJ, USA, 2013; Volume 3.
- Bassan, R.; Masciulli, A.; Intermesoli, T.; Audisio, E.; Rossi, G.; Pogliani, E.M.; Cassibba, V.; Mattei, D.; Romani, C.; Cortelezzi, A.; et al. Randomized trial of radiation-free central nervous system prophylaxis comparing intrathecal triple therapy with liposomal cytarabine in acute lymphoblastic leukemia. Haematologica 2015, 100, 786–793.
- Egusquiaguirre, S.P.; Igartua, M.; Hernández, R.M.; Pedraz, J.L. Nanoparticle delivery systems for cancer therapy: Advances in clinical and preclinical research. Clin. Transl. Oncol. 2012, 14, 83–93.
- Kim, S.; Turker, M.S.; Chi, E.Y.; Sela, S.; Martin, G.M. Preparation of multivesicular liposomes. Biochim. Biophys. Acta 1983, 728, 339–348.
- Chamberlain, M.C. Neurotoxicity of intra-CSF liposomal cytarabine (DepoCyt) administered for the treatment of leptomeningeal metastases: A retrospective case series. J. Neurooncol. 2012, 109, 143–148.
- Kim, S.; Chatelut, E.; Kim, J.C.; Howell, S.B.; Cates, C.; Kormanik, P.A.; Chamberlain, M.C. Extended CSF cytarabine exposure following intrathecal administration of DTC 101. J. Clin. Oncol. 1993, 11, 2186–2193.
- Kohn, F.R.; Malkmus, S.A.; Brownson, E.A.; Rossi, S.S.; Yaksh, T.L. Fate of the predominant phospholipid component of DepoFoamTM drug delivery matrix after intrathecal administration of sustained-release encapsulated cytarabine in rats. Drug Deliv. 1998, 5, 143–151.
- Wan, S.H.; Huffman, D.H.; Azarnoff, D.L.; Hoogstraten, B.; Larsen, W.E. Pharmacokinetics of 1-β-D-arabinofuranosylcytosine in humans. Cancer Res. 1974, 34, 392–397.
- Groothuis, D.R.; Benalcazar, H.; Allen, C.V.; Wise, R.M.; Dills, C.; Dobrescu, C.; Rothholtz, V.; Levy, R.M. Comparison of cytosine arabinoside delivery to rat brain by intravenous, intrathecal, intraventricular and intraparenchymal routes of administration. Brain Res. 2000, 856, 281–290.
- Scott-Moncrieff, J.C.R.; Chan, T.C.; Samuels, M.L.; Cook, J.R.; Coppoc, G.L.; DeNicola, D.B.; Richardson, R.C. Plasma and cerebrospinal fluid pharmacokinetics of cytosine arabinoside in dogs. Cancer Chemother. Pharmacol. 1991, 29, 13–18.
- Tardi, P.; Johnstone, S.; Harasym, N.; Xie, S.; Harasym, T.; Zisman, N.; Harvie, P.; Bermudes, D.; Mayer, L. In vivo maintenance of synergistic cytarabine: Daunorubicin ratios greatly enhances therapeutic efficacy. Leuk. Res. 2009, 33, 129–139.
- Bayne, W.F.; Mayer, L.D.; Swenson, C.E. Pharmacokinetics of CPX-351 (cytarabine/daunorubicin HCl) liposome injection in the mouse. J. Pharma. Sci. 2009, 98, 2540–2548.
- Carol, H.; Fan, M.M.; Harasym, T.O.; Boehm, I.; Mayer, L.D.; Houghton, P.; Smith, M.A.; Lock, R.B. Efficacy of CPX-351,(cytarabine: Daunorubicin) liposome injection, against acute lymphoblastic leukemia (ALL) xenograft models of the Pediatric Preclinical Testing Program. Pediatric Blood Cancer 2015, 62, 65–71.
- Dicko, A.; Kwak, S.; Frazier, A.A.; Mayer, L.D.; Liboiron, B.D. Biophysical characterization of a liposomal formulation of cytarabine and daunorubicin. Int. J. Pharma. 2010, 391, 248–259.
- Jabbour, E.; Cortes, J.E.; Giles, F.J.; O’Brien, S.; Kantarjian, H.M. Current and emerging treatment options in chronic myeloid leukemia. Cancer 2007, 109, 2171–2181.
- Shah, M.; Agarwal, B. Recent advances in management of acute myeloid leukemia (AML). Ind. J. Pediatrics 2008, 75, 831–837.
- Drugs.com. Depocyt. Available online: (accessed on 1 June 2018).
- Chamberlain, M.C.; Khatibi, S.; Kim, J.C.; Howell, S.B.; Chatelut, E.; Kim, S. Treatment of leptomeningeal metastasis with intraventricular administration of depot cytarabine (DTC 101). A phase I study. Arch. Neurol. 1993, 50, 261–264.
- Chamberlain, M.C.; Kormanik, P.; Howell, S.B.; Kim, S. Pharmacokinetics of intralumbar DTC-101 for the treatment of leptomeningeal metastases. Arch. Neurol. 1995, 52, 912–917.
- Mrugala, M.M.; Kim, B.; Sharma, A.; Johnson, N.; Graham, C.; Kurland, B.F.; Gralow, J. Phase II Study of Systemic High-dose Methotrexate and Intrathecal Liposomal Cytarabine for Treatment of Leptomeningeal Carcinomatosis From Breast Cancer. Clin. Breast Cancer 2019.
- Glantz, M.J.; Jaeckle, K.A.; Chamberlain, M.C.; Phuphanich, S.; Recht, L.; Swinnen, L.J.; Maria, B.; LaFollette, S.; Schumann, G.B.; Cole, B.F.; et al. A randomized controlled trial comparing intrathecal sustained-release cytarabine (DepoCyt) to intrathecal methotrexate in patients with neoplastic meningitis from solid tumors. Clin. Cancer Res. 1999, 5, 3394–3402.
- Phuphanich, S.; Maria, B.; Braeckman, R.; Chamberlain, M. A pharmacokinetic study of intra-CSF administered encapsulated cytarabine (DepoCyt®) for the treatment of neoplastic meningitis in patients with leukemia, lymphoma, or solid tumors as part of a phase III study. J. Neurooncol. 2007, 81, 201–208.
- Beauchesne, P.; Blonski, M.; Brissart, H. Response to intrathecal infusions of Depocyt(R) in secondary diffuse leptomeningeal gliomatosis. A case report. In vivo 2011, 25, 991–993.
- Zimm, S.; Collins, J.M.; Miser, J.; Chatterji, D.; Poplack, D.G. Cytosine arabinoside cerebrospinal fluid kinetics. Clin. Pharmacol. Ther. 1984, 35, 826–830.
- Glantz, M.J.; LaFollette, S.; Jaeckle, K.A.; Shapiro, W.; Swinnen, L.; Rozental, J.R.; Phuphanich, S.; Rogers, L.R.; Gutheil, J.C.; Batchelor, T.; et al. Randomized trial of a slow-release versus a standard formulation of cytarabine for the intrathecal treatment of lymphomatous meningitis. J. Clin. Oncol. 1999, 17, 3110–3116.
- Lassaletta, A.; Lopez-Ibor, B.; Mateos, E.; Gonzalez-Vicent, M.; Perez-Martinez, A.; Sevilla, J.; Diaz, M.A.; Madero, L. Intrathecal liposomal cytarabine in children under 4 years with malignant brain tumors. J. Neurooncol. 2009, 95, 65–69.
- Persons, S. The decline of homeopathy—the University of Iowa, 1876–1919. Bull. Hist. Med. 1991, 65, 74–87.
- Fleischhack, G.; Jaehde, U.; Bode, U. Pharmacokinetics following intraventricular administration of chemotherapy in patients with neoplastic meningitis. Clin. Pharm. 2005, 44, 1–31.
- Angst, M.S.; Drover, D.R. Pharmacology of drugs formulated with DepoFoam: A sustained release drug delivery system for parenteral administration using multivesicular liposome technology. Clin. Pharm. 2006, 45, 1153–1176.
- Bohn, J.P.; Reinstadler, V.; Pall, G.; Stockhammer, G.; Steurer, M.; Oberacher, H.; Wolf, D. Cerebrospinal Fluid Drug Concentrations and Clinical Outcome of Patients with Neoplastic Meningitis Treated with Liposomal Cytarabine. Eur. J. Drug. Metab. Pharm. 2019.
- Chen, K.T.J.; Gilabert-Oriol, R.; Bally, M.B.; Leung, A.W.Y. Recent Treatment Advances and the Role of Nanotechnology, Combination Products, and Immunotherapy in Changing the Therapeutic Landscape of Acute Myeloid Leukemia. Pharm. Res. 2019, 36, 125.
- Mayer, L.D.; Tardi, P.; Louie, A.C. CPX-351: A nanoscale liposomal co-formulation of daunorubicin and cytarabine with unique biodistribution and tumor cell uptake properties. Int. J. Nanomed. 2019, 14, 3819–3830.
- Tolcher, A.W.; Mayer, L.D. Improving combination cancer therapy: The CombiPlex((R)) development platform. Future Oncol. 2018, 14, 1317–1332.

