 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jeffrey Victor Leyton | + 12269 word(s) | 12269 | 2021-07-20 04:04:42 | | | |
| 2 | Bruce Ren | -21 word(s) | 12248 | 2021-07-21 03:26:29 | | |
Video Upload Options
Biologically-based therapies increasingly rely on the endocytic cycle of internalization and exocytosis of target receptors for cancer therapies. However, receptor trafficking pathways (endosomal sorting (recycling, lysosome localization) and lateral membrane movement) are often dysfunctional in cancer. Antibody-drug conjugates (ADCs) have revitalized the concept of targeted chemotherapy by coupling inhibitory antibodies to cytotoxic payloads. Significant advances in ADC technology and format, and target biology have hastened the FDA approval of nine ADCs (four since 2019). Although the links between aberrant endocytic machinery and cancer are emerging, the impact of dysregulated internalization processes of ADC targets and response rates or resistance have not been well studied. This is despite the reliance on ADC uptake and trafficking to lysosomes for linker cleavage and payload release.
1. Introduction
| ADC | Target | Indications and Usage 1 |
|---|---|---|
| Gemtuzumab ozogamicin (GO) | CD33 | ● Newly diagnosed and relapsed and refractory acute myeloid leukemia (AML) 2 |
| Brentuximab vedotin (BV) | CD30 | ● Hodgkin lymphoma after failure of autologous stem cell transplant (ASCT) or after failure of at least two prior multi-agent chemotherapy regimens in patients who are not eligible for ASCT ● Systemic anaplastic large cell lymphoma (ALCL) after failure of at least one prior multi-agent chemotherapy regimen |
| Inotuzumab ozogamicin (InO) | CD22 | ● Relapsed or refractory B cell precursor acute lymphoblastic lymphoma (ALL) |
| Polatuzumab vedotin (PV) | CD79b | ● In combination with bendamustine and rituximab for relapsed or refractory diffuse large B cell lymphoma after at least two prior treatments |
| Sacituzumab govitecan (SG) | Trop2 | ● Triple-negative breast cancer after at least two prior therapies for metastatic disease |
| Trastuzumab emtansine (T-DM1) | HER2 | ● Metastatic breast cancer patients who previously received trastuzumab and a taxane ● Adjuvant treatment in early breast cancer with residual invasive disease after neoadjuvant taxane- and trastuzumab-based treatment |
| Trastuzumab deruxtecan (T-DXd) | HER2 | ● Unresectable or metastatic breast cancer after two or more prior anti-HER2-based regimens in the metastatic setting |
| Enfortumab vedotin (EV) |
Nectin-4 | ● Locally advanced or metastatic urothelial cancer after a PD-1 3 or PD-L1 inhibitor, and platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally, advanced or metastatic setting |
| Belantamab mafodotin (BM) | BCMA | ● Relapsed or refractory multiple myeloma after at least four prior therapies including an anti-CD38 mAb, a proteasome inhibitor, and an immunomodulatory agent |
2. Relevant Endocytic Pathways for Currently Approved ADCs
2.1. Clathrin-Mediated Endocytosis
2.2. Clathrin-Independent Endocytosis
2.2.1. Caveolae-Mediated Endocytosis
2.2.2. CLIC/GEEC Endocytosis
2.2.3. Macropinocytosis
3. Target Antigens for Approved ADCs and Their Endocytosis Characteristics
3.1. CD33
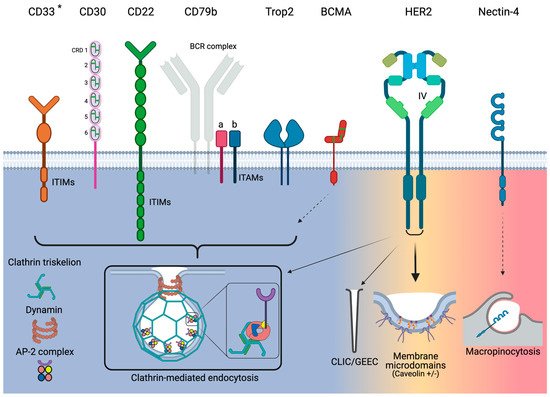
3.2. ADC/Ab-CD33 Endocytosis
| Receptor | Pathway | Activity | Association with ADC Efficacy/Resistance |
|---|---|---|---|
| CD33 | CME | Poor | ● AML patients who do not respond to GO have been linked to poor receptor internalization |
| CD30 | CME | Poor | ● Undergoes significant shedding from cell surface |
| CD22 | CME | Good | ● Fast endocytosis activates intracellular pools, which replenish the level of CD22 expression |
| CD79b | CME | Good | ● Due to rapid internalization and trafficking to lysosomes, patients will most likely respond to PV treatment |
| Trop2 | CME | Good | ● Strong preclinical data link internalization to efficacy |
| BCMA | Insufficient information | Good | ● Insufficient information |
| HER2 | Clathrin-independent (caveolae +/−) | Poor | ● Poor internalization linked with poor clinical outcomes ● Dysregulation of the endocytotic machinery has been linked to resistance in preclinical models ● Novel strategies such as induced HER2 crosslinking to improve endocytosis are currently in clinical testing |
| Nectin-4 | Macropinocytosis | Good | ● Insufficient information |
3.3. CD30
3.4. ADC/Ab-CD30 Endocytosis
3.5. CD22
3.6. ADC/Ab-CD22 Endocytosis
3.7. CD79b
3.8. ADC/Ab-CD79b Endocytosis
3.9. Trop2
3.10. ADC/Ab-Trop2 Endocytosis
3.11. BCMA
3.12. ADC/Ab-BCMA Endocytosis
3.13. HER2 and CME
3.13.1. HER2 and Clathrin-Independent Endocytosis
HER2 and Caveolae-Mediated Endocytosis
HER2 and CLIC/GEEC Endocytosis
3.14. Ab/ADC-HER2 Endocytosis
3.15. Nectin-4
References
- Clinicaltrials.gov. Keywords: Antibody-Drug Conjugates. Available online: (accessed on 30 April 2021).
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab vedotin in relapsed or refractory diffuse large B-cell lymphoma. J. Clin. Oncol. 2020, 38, 155–165.
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-programmed death 1/programmed death ligand 1 therapy. J. Clin. Oncol. 2019, 37, 2592–2600.
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzmab deruxtecan in previously treated HER2-positive breast cancer. N. Engl. Med. 2020, 382, 610–621.
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N. Engl. Med. 2019, 380, 741–751.
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. mAbs 2013, 5, 13–21.
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169.
- Leyton, J.V. Improving receptor-mediated intracellular access and accumulation of antibody therapeutics—The tale of HER2. Antibodies 2020, 9, 32.
- Lambert, J.M.; Morris, C.Q. Antibody-drug conjugates (ADCs) for personalized treatment of solid tumors: A review. Adv. Ther. 2017, 34, 1015–1035.
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-drug conjugates: A comprehensive review. Mol. Cancer Res. 2020, 18, 3–19.
- Sievers, E.L.; Senter, P.D. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013, 64, 15–29.
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537.
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016, 17, e254–e262.
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody-drug conjugates for cancer therapy. Molecules 2020, 25, 4764.
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug. Discov. 2017, 16, 315–337.
- Polakis, P. Antibody-drug conjugates for cancer therapy. Pharmacol. Rev. 2016, 68, 3–19.
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs 2016, 8, 659–671.
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351.
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell. 2018, 9, 33–46.
- Ducry, L.; Stump, B. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjug. Chem. 2010, 21, 5–13.
- Kostova, V.; Desos, P.; Starck, J.-B.; Kotschy, A. The chemistry behind ADCs. Pharmaceuticals 2021, 14, 442.
- Garcia-Alonso, S.; Ocaña, A.; Pandiella, A. Resistance to antibody-drug conjugates. Cancer Res. 2018, 78, 2159–2165.
- Collins, D.M.; Bossenmaier, B.; Kollmorgen, G.; Niederfellner, G. Acquired resistance to antibody-drug conjugates. Cancers 2019, 11, 394.
- Chalouni, C.; Doll, S. Fate of antibody-drug conjugates in cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 20.
- Kalim, M.; Chen, J.; Wang, S.; Ullah, S.; Liang, K.; Ding, Q.; Chen, S.; Zhan, J. Intracellular trafficking of new anticancer therapeutics: Antibody-drug conjugates. Drug Des. Dev. Ther. 2019, 11, 2265–2276.
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902.
- Kaksonen, M.; Roux, A. mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326.
- Mosesson, Y.; Mills, G.B.; Tarden, Y. Derailed endocytosis: An emerging feature of cancer. Nat. Rev. Cancer 2008, 8, 835–850.
- Abella, J.V.; Park, M. Breakdown of endocytosis in the oncogenic activation of receptor tyrosine kinases. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E973–E984.
- Mahalingaiah, P.K.; Ciurlionis, R.; Durbin, K.R.; Yeager, R.L.; Philip, B.K.; Bawa, B.; Mantena, S.R.; Enright, B.P.; Liguori, M.J.; Van Vleet, T.R. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther. 2019, 200, 110–125.
- Praefcke, G.J.K.; McMahon, H.T. The dynamin superfamily: Universal membrane tabulation and fission molecules? Nat. Rev. Mol. Cell Biol. 2004, 5, 133–147.
- Scherer, P.E.; Okamoto, T.; Chun, M.; Nishimoto, I.; Lodish, H.F.; Lisanti, M.P. Identification, sequence, and expression of caveolin-2 defines a ceveolin gene family. Proc. Natl. Acad. Sci. USA 1996, 93, 131–135.
- Tang, Z.; Scherer, P.E.; Okamoto, T.; Song, K.; Chu, C.; Kohtz, D.S.; Nishimoto, I.; Lodish, H.F.; Lisanti, M.P. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J. Biol. Chem. 1996, 271, 2255–2261.
- Feron, O.; Belhassen, L.; Kobzik, L.; Smith, T.W.; Kelly, R.A.; Michel, T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J. Biol. Chem. 1996, 271, 22810–22814.
- Couet, J.; Li, S.; Okamoto, T.; Ikezu, T.; Lisanti, M.P. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J. Biol. Chem. 1997, 272, 6525–6533.
- Sargiacomo, M.; Scherer, P.E.; Tang, Z.; Kubler, E.; Song, K.S.; Sanders, M.C.; Lisanti, M.P. Oligomeric structure of caveolin: Implications for caveolae membrane organization. Proc. Natl. Acad. Sci. USA 1995, 92, 9407–9411.
- Shajahan, A.N.; Timblin, B.K.; Sandoval, R.; Tiruppathi, C.; Malik, A.B.; Minshall, R.D. Role of Src-induced dynamin-2 phophorylation in caveolae-mediated endocytosis in endothelial cells. J. Biol. Chem. 2004, 279, 20392–20400.
- Senju, Y.; Itoh, Y.; Takano, K.; Hamada, S.; Suetsugu, S. Essential role of PACSIN2/syndapin-II caveolae membrane sculpting. J. Cell Sci. 2011, 124, 2032–2040.
- Parton, R.G.; Tillu, V.A.; Collins, B.M. Caveolae. Curr. Biol. 2018, 28, R402–R405.
- Kirkham, M.; Parton, P.G. Clathrin-independent endocytosis: New insights into caveolae and non-caveolar lipid raft carriers. Biochim. Biophys. Acta 2005, 1746, 349–363.
- Pelkmans, L.; Burli, T.; Zerial, M.; Helenius, A. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell 2004, 118, 767–780.
- Moon, H.; Lee, C.S.; Inder, K.L.; Sharma, S.; Choi, E.; Black, D.M.; Cao, K.-A.L.; Winterford, C.; Coward, J.I.; Lint, M.T.; et al. PTRF/cavin-1 neutralizes non-caveolar caveolin-1 microdomains in prostate cancer. Oncogene 2014, 33, 3561–3570.
- Corn, P.G.; Thompson, T.C. Identification of a novel prostate cancer biomarker, caveolin-1: Implications and potential clinical benefit. Cancer Manag. Res. 2010, 2, 111–122.
- Ferreira, A.P.A.; Boucrot, E. Mechanisms of carrier formation during clathrin-independent endocytosis. Trends Cell Biol. 2018, 28, 188–200.
- Howes, M.T.; Kirkham, M.; Riches, J.; Cortese, K.; Walser, P.J.; Simpson, F.; Hill, M.M.; Jones, A.; Lundmark, R.; Lindsay, M.R.; et al. Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J. Cell Biol. 2010, 190, 675–691.
- Sabharanjak, S.; Pranav, S.; Paton, R.G.; Mayor, S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell 2002, 2, 411–423.
- Chadda, R.; Howes, M.T.; Plowman, S.J.; Hancock, J.F.; Parton, R.G.; Mayor, S. Cholesterol-sensitive cdc42 activation regulates actin polymerization for endocytosis via the GEEC pathway. Traffic 2007, 8, 702–717.
- Yarar, D.; Waterman-Storer, C.M.; Schmid, S.L. SNX9 couples actin assembly to phosphoinositide signals and is required for membrane remodeling during endocytosis. Dev. Cell 2007, 13, 43–56.
- Lundmark, R.; Doherty, G.J.; Howes, M.T.; Cortese, K.; Vallis, Y.; Parton, R.G.; McMahon, H.T. The GTPase-activating protein GRAF1 regulates the CLIC/GEEC endocytic pathway. Curr. Biol. 2008, 18, 1802–1808.
- Knaus, U.G.; Wang, Y.; Reilly, A.M.; Warnock, D.; Jackson, J.H. Structural requirement for PAK activation by Rac GTPases. J. Biol. Chem. 1998, 273, 21512–21518.
- Amyere, M.; Payrastre, B.; Krause, U.; Van der Smissen, P.; Veithen, A.; Courtoy, P.J. Constitutive macropinocytosis in oncogene-transformed fibroblasts depends on sequential permanent activation of phosphoinositide 3-kinase and phospholipase C. Mol. Biol. Cell 2000, 11, 3453–3467.
- Veithen, A.; Cupers, P.; Baudhuin, P.; Courtoy, P.J. v-Src induces constitutive macropinocytosis in rat fibroblasts. J. Cell Sci. 1996, 109, 2005–2012.
- Gao, Y.S.; Hubbert, C.C.; Lu, J.; Lee, Y.-S.; Lee, J.-Y.; Yao, T.-P. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol. Cell Biol. 2007, 27, 8637–8647.
- Liberali, P.; Kakkonen, E.; Turacchio, G.; Valente, C.; Spaar, A.; Perinetti, G.; Bockmann, R.A.; Corda, D.; Colanzi, A.; Marjomaki, V.; et al. The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J. 2008, 27, 970–981.
- Grimmer, S.; van Deurs, B.; Sandvig, K. Membrane ruffling and macropinocytosis in A431 cells require cholesterol. J. Cell Sci. 2002, 115, 2953–2962.
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153.
- Verbrugge, A.; Meyaard, L. Signaling by ITIM-bearing receptors. Curr. Immunol. Rev. 2005, 1, 201–212.
- Taylor, V.C.; Buckley, C.D.; Douglas, M.; Simmons, D.L.; Freeman, S.D. The myeloid-specific sialic acid-binding receptor, CD33, associates with the protein-tyrosine phosphatases, SHP-1 and SHP-2. J. Biol. Chem. 1999, 274, 11505–11512.
- Paul, S.P.; Taylor, L.S.; Stansbury, E.K.; McVicar, D.W. Myeloid specific human CD33 is an inhibitory receptor with differential ITIM function in recruiting the phosphatases SHP-1 and SHP-2. Blood. 2000, 96, 483–490.
- Hernandez-Caselles, T.; Corral-San Miguel, R.; Ruiz-Alcaraz, J.R.; Garcia-Peñarrubia, P. CD33 (Siglec-3) inhibitory function: Role in the NKG2D/DAP10 activating pathway. J. Immunol. Res. 2019, 2019, 6032141.
- Walter, R.B.; Raden, B.W.; Kamikura, D.M.; Cooper, J.A.; Bernstein, I.D. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin-induced cytotoxicity. Blood 2005, 105, 1295–1302.
- Walter, R.B.; Raden, B.W.; Zeng, R.; Hausermann, P.; Bernstein, I.D.; Cooper, J.A. ITIM-dependent endocytosis of CD33-related siglecs: Role of intracellular domain, tyrosine phosphorylation, and the tyrosine phosphatases, Shp1 and Shp2. J. Leukoc. Biol. 2008, 83, 200–211.
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266.
- Van der Velden, V.H.J.; te Marvelde, J.G.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; van Dongen, J.J.M. Targeting of CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204.
- Caron, P.C.; Schwartz, M.A.; Co, M.S.; Queen, C.; Finn, R.D.; Graham, M.C.; Divgi, C.R.; Larson, S.M.; Scheinberg, D.A. Murine and humanized constructs of monoclonal antibody M195 (anti-CD33) for the therapy of acute myelogenous leukemia. Cancer 1994, 73, 1049–1056.
- Jedema, I.; Barge, R.M.Y.; van der Velden, V.H.J.; Nijmeijer, B.A.; van Dongen, J.J.M.; Willemze, R.; Falkenburg, J.H.F. Internalization and cell cycle-dependent killing of leukemic cells by gemtuzumab ozogamicin: Rationale for efficacy in CD33-negative malignancies with endocytic capacity. Leukemia 2004, 18, 316–325.
- Paubelle, E.; Marceau, A.; Zylbersztejn, F.; Dussiot, M.; Cruz Moura, I.; Cornillet-Lefebvre, P.; Delaunay, J.; Burnett, A.K.; Castaigne, S.; Guardolia, P.; et al. HFE gene mutation status predicts response to gemtuzumab ozogamicin in AML. Blood 2015, 126, 1307.
- Orr, S.J.; Morgan, N.M.; Elliott, J.; Burrows, J.F.; Scott, C.J.; McVicar, D.W.; Johnston, J.A. CD33 responses are blocked by SOC3 through accelerated proteasomal-mediated turnover. Blood 2007, 109, 1061–1068.
- Smith, C.A.; Gruss, H.J.; Davis, T.; Anderson, D.; Farrah, T.; Baker, E.; Sutherland, G.R.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A. CD30 antigen, a marker for Hodgkin’s lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell 1993, 73, 1349–1360.
- Mukai, Y.; Nakamura, T.; Yoshikawa, M.; Yoshioka, Y.; Tsunoda, S.; Nakagawa, S.; Yamagata, Y.; Tsutsumi, Y. Solution of the structure of the TNF-TNFR2 complex. Sci. Signal. 2010, 3, ra83.
- Siegel, R.M.; Frederiksen, J.K.; Zacharias, D.A.; Chan, F.K.; Johnson, M.; Lynch, D.; Tsien, R.Y.; Lenardo, M.J. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 2000, 288, 2354–2357.
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000, 288, 2351–2354.
- Park, Y.C.; Burkitt, V.; Villa, A.R.; Tong, L.; Wu, H. Structural basis for self-association and receptor recognition of human TRAF2. Nature 1999, 398, 533–538.
- Van der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603.
- Gruss, H.J.; Boiani, N.; Williams, D.E.; Armitage, R.J.; Smith, C.A.; Goodwin, R.G. Pleiotropic effects of the CD30 ligand on CD30-expressing cells and lymphoma cell lines. Blood 1994, 83, 2045–2056.
- Hansen, H.P.; Dietrich, S.; Kisseleva, T.; Mokros, T.; Mentlein, R.; Lange, H.H.; Murphy, G.; Lemke, H. CD30 shedding from Karpas 299 lymphoma cells is mediated by TNF-α-converting enzyme. Immunology 2000, 165, 6703–6709.
- Eichenauer, D.A.; Simhadri, V.L.; von Strandmann, E.P.; Ludwig, A.; Matthews, V.; Reiners, K.S.; von Tresckow, B.; Saftig, P.; Rose-John, S.; Engert, A.; et al. ADAM10 inhibition of human CD30 shedding increases specificity of targeted immunotherapy in vitro. Cancer Res. 2007, 67, 332–338.
- Hansen, H.P.; Recke, A.; Reineke, U.; Von Tresckow, B.; Borchmann, P.; Von Strandmann, E.P.; Lange, H.; Lemke, H.; Engert, A. The ectodomain shedding of CD30 is specifically regulated by peptide motifs in its cysteine-rich domains 2 and 5. FASEB J. 2004, 18, 893–905.
- Borchmann, P.; Tremi, J.F.; Gottstein, C.; Schnell, R.; Staak, O.; Zhang, H.; Davis, T.; Keier, T.; Diehl, V.; Graziano, R.F. The human anti-CD30 antibody 5F11 shows in vitro and in vivo activity against malignant lymphoma. Blood 2003, 102, 3737–3742.
- Matthey, B.; Borchmann, P.; Schnell, R.; Tawadros, S.; Lange, H.; Huhn, M.; Klimka, A.; Tur, M.K.; Barth, S.; Engert, A.; et al. Metalloproteinase inhibition augments antitumor efficacy of the anti-CD30 immunotoxin Ki-3 (scFv)-ETA′ against human lymphomas in vivo. Int. J. Cancer 2004, 111, 568–574.
- Wall, L.; Talbot, D.C.; Bradbury, P.; Jodrell, D.I. A phase I and pharmacological study of the matrix metalloproteinase inhibitor BB-3644 in patients with solid tumors. Br. J. Cancer 2004, 90, 800–804.
- Hansen, H.P.; Paes Leme, A.F.; Hallek, M. Role of ADAM10 as a CD30 sheddase in classical Hodgkin lymphoma. Front. Immunol. 2020, 11, 398.
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999, 97, 133–144.
- Schirrmann, T.; Steinwand, M.; Wezler, X.; ten Haaf, A.; Tur, M.K.; Barth, S. CD30 as a therapeutic target for lymphoma. BioDrugs 2014, 28, 181–209.
- Sutherland, M.S.K.; Sanderson, R.J.; Gordon, K.A.; Andreyka, J.; Cerveny, C.G.; Yu, C.; Lewis, T.S.; Meyer, D.L.; Zabinski, R.F.; Doronina, S.O.; et al. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J. Biol. Chem. 2006, 281, 10540–10547.
- Okeley, N.M.; Miyamoto, J.B.; Zhang, X.; Sanderson, R.J.; Benjamin, D.R.; Sievers, E.L.; Senter, P.D.; Alley, S.C. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin. Cancer Res. 2010, 16, 888–897.
- Lanza, F.; Maffini, E.; Rondoni, E.; Faini, A.C.; Malavasi, F. CD22 expression in B-cell acute lymphoblastic leukemia: Biological significance and implications for inotuzumab therapy in adults. Cancers 2020, 12, 303.
- Ereño-Orbea, J.; Sicard, T.; Cui, H.; Mazhab-Jafari, M.T.; Benlekbir, S.; Guarné, A.; Rubinstein, J.L.; Julien, J.-P. Molecular basis of human CD22 function and therapeutic targeting. Nat. Commun. 2017, 8, 764.
- O’Reilly, M.K.; Collins, B.E.; Han, S.; Liao, L.; Rillahan, C.; Kitov, P.I.; Bundle, D.R.; Paulson, J.C. Bifunctional CD22 ligands use multimeric immunoglobulins as protein scaffolds in assembly of immune complexes of B cells. J. Am. Chem. Soc. 2008, 130, 7736–7745.
- Collins, B.E.; Blixt, O.; Han, S.; Duong, B.; Li, H.; Nathan, J.K.; Bovin, N.; Paulson, J.C. High-affinity ligand probes of CD22 overcome the threshold set by cis ligands to allow for binding, endocytosis, and killing of B cells. J. Immunol. 2006, 177, 2994–3003.
- John, B.; Herrin, B.R.; Raman, C.; Wang, Y.; Bobbitt, K.R.; Brody, B.A.; Justement, L.B. The B cell coreceptor CD22 associates with AP50, a clathrin-coated pit adapter protein, via tyrosine-dependent interaction. J. Immunol. 2003, 170, 3534–3543.
- O’Reilly, M.K.; Tian, H.; Paulson, J.C. CD22 is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B cells. J. Immunol. 2011, 186, 1554–1563.
- Shan, D.; Press, O.W. Constitutive endocytosis and degradation of CD22 by human B cells. J. Immunol. 1995, 154, 4466–4475.
- Carnahan, J.; Wang, P.; Kendall, R.; Chen, C.; Hu, S.; Boone, T.; Juan, T.; Talvenheimo, J.; Montrestruque, S.; Sun, J.; et al. Epratuzumab, a humanized monoclonal antibody targeting CD22: Characterization of in vitro properties. Clin. Cancer Res. 2003, 9, 3982S–3990S.
- Du, X.; Beers, R.; Fitzgerald, D.J.; Pastan, I. Differential cellular internalization of anti-CD19 and -CD22 immunotoxins results in different cytotoxic activity. Cancer Res. 2008, 68, 6300–6305.
- Fingrut, W.; Davis, W.; McGinnis, E.; Dallas, K.; Ramadan, K.; Merkeley, H.; Leitch, H.; Mourad, Y.A.; Cassaday, R.D.; Ross, C. Reevaluating Patient Eligibility for Inotuzumab Ozogamicin Based on CD22 Expression: Is Dim Expression Sufficient? Curr. Oncol. 2021, 28, 252–259.
- Horvat, M.; Zadnik, V.; Juznic Setina, T.; Boltezar, L.; Golicnik, J.P.; Novakovic, S.; Jezersek Novakovic, B. Diffuse large B-cell lymphoma: 10 years’ real-world clinical experience with rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisolone. Oncol. Lett. 2018, 15, 3602–3609.
- Pfeifer, M.; Zheng, B.; Erdmann, T.; Koeppen, H.; McCord, R.; Grau, M.; Staiger, A.; Chai, A.; Sandmann, T.; Madle, H.; et al. Anti-CD22 and anti-CD79b antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia 2015, 29, 1578–1586.
- Chu, P.G.; Arber, D.A. CD79: A review. Appl. Immunohistochem. Mol. Morphol. 2001, 9, 97–106.
- Busman-Sahay, K.; Drake, L.; Sitaram, A.; Marks, M.; Drake, J.R. Cis and trans regulatory mechanism control AP2-mediated B cell receptor endocytosis via select tyrosine-based motifs. PLoS ONE 2013, 8, e54938.
- Hou, P.; Araujo, E.; Zhao, T.; Zhang, M.; Massenburg, D.; Veselits, M.; Doyle, C.; Dinner, A.R.; Clark, M.R. B cell antigen receptor signaling and internalization are mutually exclusive events. PLoS Biol. 2006, 4, e200.
- Drake, J.R.; Lewis, T.A.; Condon, K.B.; Mitchell, R.N.; Webster, P. Involvement of MIIC-like late endosomes in B cell receptor-mediated antigen processing in murine B cells. J. Immunol. 1999, 162, 1150–1155.
- Salisbury, J.L.; Condeelis, J.S.; Satir, P. Role of coated vesicles, microfilaments, and calmodulin in receptor-mediated endocytosis by cultured B lymphoblastoid cells. J. Cell Biol. 1980, 87, 132–141.
- Kim, J.H.; Rutan, J.A.; Vilen, B.J. The transmembrane tyrosine of μ-heavy chain is required for BCR destabilization and entry of antigen into clathrin-coated vesicles. Int. Immunol. 2007, 19, 1403–1412.
- Caballero, A.; Katkere, B.; Wen, X.-Y.; Drake, L.; Nashar, T.O.; Drake, J.R. Functional and structural requirements for the internalization of distinct BCR-ligand complexes. Eur. J. Immunol. 2006, 36, 3131–3145.
- Stoddart, A.; Jackson, A.P.; Brodsky, F.M. Plasticity of B cell receptor internalization upon conditional depletion of clathrin. Mol. Biol. Cell 2005, 16, 2339–2348.
- Jang, C.; Machtaler, S.; Matsuuchi, L. The role of Ig-alpha/beta in B cell antigen receptor internalization. Immunol. Lett. 2010, 134, 75–82.
- Luisiri, P.; Lee, Y.J.; Eisfelder, B.J.; Clark, M.R. Cooperativity and segregation of function within the Ig-alpha/beta heterodimer of the B cell antigen receptor complex. J. Biol. Chem. 1996, 271, 5158–5163.
- Patel, P.J.; Neuberger, M.S. Antigen presentation by the B cell antigen receptor is driven by the alpha/beta sheath and occurs independently of its cytoplasmic tyrosines. Cell 1993, 74, 939–946.
- Siemasko, K.; Eisfelder, B.J.; Stebbins, C.; Kabak, S.; Sant, A.J.; Song, W.; Clark, M.R. Ig alpha and Ig beta are required for efficient trafficking to late endosomes and to enhance antigen presentation. J. Immunol. 1999, 162, 6518–6525.
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signaling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92.
- Stoddart, A.; Dykstra, M.L.; Brown, B.K.; Song, W.; Pierce, S.K.; Brodsky, F.M. Lipid rafts unite signaling cascades with clathrin to regulate BCR internalization. Immunity 2002, 17, 451–462.
- Okazaki, M.; Luo, Y.; Yoshida, M.; Seon, B.K. Three new monoclonal antibodies that define a unique antigen associated with prolymphocytic leukemia/non-Hodgkin’s lymphoma and are effectively internalized after binding to the cell surface antigen. Blood 1993, 81, 84–94.
- Polson, A.G.; Yu, S.-F.; Elkins, K.; Zheng, B.; Clark, S.; Ingle, G.S.; Slaga, D.S.; Giere, L.; Du, C.; Tan, C.; et al. Antibody-drug conjugate targeted to CD79 for the treatment of non-Hodgkin lymphoma. Blood 2007, 110, 616–623.
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; de Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-drug conjugates for the treatment of non-Hodgkin’s lymphoma: Target and linker-drug selection. Cancer Res. 2009, 69, 2358–2364.
- Dornan, D.; Bennett, F.; Chen, Y.; Dennis, M.; Eaton, D.; Elkins, K.; French, D.; Go, M.A.T.; Jack, A.; Junutula, J.R.; et al. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood 2009, 114, 2721–2729.
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407.
- Gazumyan, A.; Reichlin, A.; Nussenzweig, M.C. Ig beta tyrosine residues contribute to the control of B cell receptor signaling by regulating receptor internalization. J. Exp. Med. 2006, 203, 1785–1794.
- Lenart, S.; Lenart, P.; Smarda, J.; Remsik, J.; Soucek, K.; Benes, P. Trop2: Jack of all trades, master of none. Cancers 2020, 12, 3328.
- Trerotola, M.; Cantanelli, P.; Guerra, E.; Tripaldi, R.; Aloisi, A.L.; Bonasera, V.; Laffanzio, R.; de Lange, R.; Weidle, U.H.; Piantelli, M.; et al. Upregulation of Trop-2 quantitatively stimulates human cancer growth. Oncogene 2013, 32, 222–233.
- Strop, P.; Tran, T.-T.; Dorywalska, M.; Delaria, K.; Dushin, R.; Wong, O.K.; Ho, W.-H.; Zhou, D.; Wu, A.; Kraynov, E.; et al. RN927C, a site-specific Trop-2 antibody-drug conjugate (ADC) with enhanced stability, is highly efficacious in preclinical solid tumor models. Mol. Cancer Ther. 2016, 15, 2698–2708.
- Wanger, T.M.; Dewitt, S.; Collins, A.; Maitland, N.J.; Poghosyan, Z.; Knauper, V. Differential regulation of TROP2 release by PKC isoforms through vesicles and ADAM17. Cell Signal. 2015, 27, 1325–1335.
- Stoyanova, T.; Goldstein, A.S.; Cai, H.; Drake, J.M.; Huang, J.; Witte, O.N. Regulated proteolysis of Trop2 drives epithelial hyperplasia and stem cell self-renewal via β-catenin signaling. Genes Dev. 2012, 26, 2271–2285.
- Fu, Y.; Hua, P.; Lou, Y.; Li, Z.; Jia, M.; Jing, Y.; Cai, M.; Wang, H.; Tong, T.; Gao, J. Mechanistic insights into Trop2 clustering on lung cancer cell membranes revealed by super-resolution imaging. ACS Omega 2020, 5, 32456–32465.
- Mooren, O.L.; Galletta, B.J.; Cooper, J.A. Roles for acting assembly in endocytosis. Annu. Rev. Biochem. 2012, 81, 661–686.
- Pavsic, M.; Ilc, G.; Vidmar, T.; Plavec, J.; Lenarcic, B. The cytosolic tail of the tumor marker protein Trop2—A structural switch triggered by phosphorylation. Sci. Rep. 2015, 5, 10324.
- Shvartsur, A.; Bonavida, B. Trop2 and its overexpression in cancers: Regulation and clinical/therapeutic implications. Genes Cancer 2015, 6, 84–105.
- Lin, J.-C.; Wu, Y.-Y.; Wu, J.-Y.; Lin, T.-C.; Wu, C.-T.; Chang, Y.-L.; Jou, Y.-S.; Hong, T.-M.; Yang, P.-C. TROP2 is epigenetically inactivated and modulates IGF-1R signaling in lung adenocarcinoma. EMBO Mol. Med. 2012, 4, 472–485.
- Zhang, K.; Jones, L.; Lim, S.; Maher, C.A.; Adkins, D.; Lewis, J.; Kimple, R.J.; Fertig, E.J.; Chung, C.H.; Herrlich, A.; et al. Loss of Trop2 causes ErbB3 activation through a neuregulin-1-dependent mechanism in the mesenchymal subtype of HNSCC. Oncotarget 2014, 5, 9281–9294.
- Stein, R.; Chen, S.; Sharkey, R.M.; Goldenberg, D.M. Murine monoclonal antibodies raised against human non-small cell carcinoma of the lung: Specificity and tumor targeting. Cancer Res. 1990, 50, 1330–1336.
- Shih, L.B.; Xuan, H.; Aninipot, R.; Stein, R.; Goldenberg, D.M. In vitro and in vivo reactivity of an internalizing antibody, RS7, with human breast cancer. Cancer Res. 1995, 55, 5857–5863.
- Stein, R.; Basu, A.; Chen, S.; Shih, L.B.; Goldenberg, D.M. Specificity and properties of mAb rS7-3G11 and the antigen defined by this pancarcinoma monoclonal antibody. Int. J. Cancer. 1993, 55, 938–946.
- Ambrogi, F.; Fornili, M.; Boracchi, P.; Trerotola, M.; Relli, V.; Simeone, P.; La Sorda, R.; Lattanzio, R.; Querzoli, P.; Pedriali, M.; et al. Trop-2 is a determinant of breast cancer survival. PLoS ONE 2014, 9, e96993.
- Lin, H.; Zhang, H.; Wang, J.; Lu, M.; Zheng, F.; Wang, C.; Tang, X.; Xu, N.; Chen, R.; Zhang, D.; et al. A novel human Fab antibody for Trop2 inhibits breast cancer growth in vitro and in vivo. Int. J. Cancer 2014, 134, 1239–1249.
- Nishimura, T.; Mitsunaga, M.; Sawada, R.; Saruta, M.; Kobayashi, H.; Matsumoto, N.; Kanke, T.; Yanai, H.; Nakamura, K. Photoimmunotherapy targeting biliary-pancreatic cancer with humanized anti-TROP2 antibody. Cancer Med. 2019, 8, 7781–7792.
- Ripani, E.; Sacchetti, A.; Corda, D.; Alberti, S. Human Trop-2 is a tumor-associated calcium signal transducer. Int. J. Cancer 1998, 76, 671–676.
- Moon, S.-J.; Govindan, S.V.; Cardillo, T.M.; D’Souza, C.A.; Hansen, H.J.; Goldenberg, D.M. Antibody conjugates of 7-ethyl-10-hydroxycamptothecin (SN-38) for targeted cancer chemotherapy. J. Med. Chem. 2008, 51, 6916–6926.
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; Trisal, P.; Goldenberg, D.M. Humanized anti-Trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers: Preclinical studies in human cancer xenograft models and monkeys. Clin. Cancer Res. 2011, 17, 3157–3169.
- Coquery, C.M.; Erickson, L.D. Regulatory roles of the tumor necrosis factor receptor BCMA. Crit. Rev. Immunol. 2013, 32, 287–305.
- Hatzoglou, A.; Roussel, J.; Bourgeade, M.-F.; Rogier, E.; Madry, C.; Inoue, J.; Devergne, O.; Tsapis, A. TNF receptor family member BCMA (B cell maturation) associates with TNF receptor-associated factor (TRAF)1, TRAF2, and TRAF3 and activates NF-κB, Elk-1, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase. J. Immunol. 2000, 165, 1322–1330.
- Madry, C.; Laabi, Y.; Callebaut, I.; Roussel, J.; Hatzoglou, A.; Le Coniat, M.; Mornon, J.P.; Berger, R.; Tsapis, A. The characterization of murine BCMA gene defines it as a new member of the tumor necrosis factor receptor superfamily. Int. Immunol. 1998, 10, 1693–1702.
- Chiu, A.; Xu, W.; He, B.; Dillon, S.R.; Gross, J.A.; Sievers, E.; Qiao, X.; Santini, P.; Hyjek, E.; Lee, J.W.; et al. Hodgkin lymphoma cells express TAC1 and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood 2007, 109, 729–739.
- Yurchenko, M.; Sidorenko, S.P. Hodgkin’s lymphoma: The role of cell surface receptors in regulation of tumor cell fate. Exp. Oncol. 2010, 32, 214–223.
- Novak, A.J.; Darce, J.R.; Arendt, B.K.; Harder, B.; Henderson, K.; Kindsvogel, W.; Gross, J.A.; Greipp, P.R.; Jelinek, D.F. Expression of BCMA, TAC1, and BAFF-R in multiple myeloma: A mechanism for growth and survival. Blood 2004, 103, 689–694.
- Schwaller, J.; Schneider, P.; Mhawech-Fauceglia, P.; McKee, T.; Myet, S.; Tschopp, M.T.; Donze, O.; Le Gal, F.A.; Huard, B. Neutrophil-derived APRIL concentrated in tumor lesions by proteoglycans correlates with human B-cell lymphoma aggressiveness. Blood 2007, 109, 331–338.
- He, B.; Chadburn, A.; Jou, E.; Schattner, E.J.; Knowles, D.M.; Cerutti, A. Lymphoma B cells evade apoptosis through the TNF family members BAFF/BLyS and APRIL. J. Immunol. 2004, 172, 3268–3279.
- Yu, B.; Jiang, T.; Liu, D. BCMA-targeted immunotherapy for multiple myeloma. J. Hematol. Oncol. 2020, 13, 125.
- Wei, J.; Han, X.; Bo, J.; Han, W. Target selection for CAR-T therapy. J. Hematol. Oncol. 2019, 12, 62.
- Eckhert, E.; Hewitt, R.; Liedtke, M. B-cell maturation antigen directed monoclonal antibody therapies for multiple myeloma. Immunotherapy 2019, 11, 801–811.
- Tai, Y.-T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236.
- Huang, H.-W.; Chen, C.-H.; Lin, C.-H.; Wong, C.-H.; Lin, K.-I. B-cell maturation antigen is modified by a single N-glycan chain that modulates ligand binding and surface retention. Proc. Natl. Acad. Sci. USA 2013, 110, 10928–10933.
- Gravestein, L.A.; Borst, J. Tumor necrosis factor receptor family members in the immune system. Semin. Immunol. 1998, 10, 423–434.
- Ji, W.; Li, Y.; Wan, T.; Wang, J.; Zhang, H.; Chen, H.; Min, W. Both internalization and AIP1 association are required for tumor necrosis factor receptor-2-mediated JNK signaling. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2271–2279.
- D’Alessio, A.; Al-Lamki, R.S.; Bradley, J.R.; Pober, J.S. Caveolae participate in tumor necrosis factor receptor 1 signaling and internalization in a human endothelial cell line. Am. J. Pathol. 2005, 166, 1273–1282.
- Sanchez, E.; Li, M.; Kitto, A.; Li, J.; Wang, C.S.; Kirk, D.T.; Yellin, O.; Nichols, C.M.; Dreyer, M.P.; Ahles, C.P.; et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br. J. Haematol. 2012, 158, 727–738.
- Ghermezi, M.; Li, M.; Vardanyan, S.; Harutyunyan, N.M.; Gottlieb, J.; Berenson, A.; Spektor, T.M.; Andreu-Vieyra, C.; Petraki, S.; Sanchez, E.; et al. Serum B-cell maturation antigen: A novel biomarker to predict outcomes for multiple myeloma patients. Haematologica 2017, 102, 785–795.
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.-H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rubsamen, H.; et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333.
- Figueroa-Vazquez, V.; Ko, J.; Breunig, C.; Baumann, A.; Giesen, N.; Palfi, A.; Muller, C.; Lutz, C.; Hechler, T.; Kulke, M.; et al. HDP-101, an anti-BCMA antibody-drug conjugate, safely delivers amanitin to induce cell death in proliferating and resting multiple myeloma cells. Mol. Cancer Ther. 2021, 20, 367–378.
- Gras, M.P.; Laabi, Y.; Linares-Cruz, G.; Blondel, M.O.; Rigaut, J.P.; Brouet, J.C.; Leca, G.; Haguenauer-Tsapis, R.; Tsapis, A. BCMAp: An integral membrane protein in the Golgi apparatus of human mature B lymphocytes. Int. Immunol. 1995, 7, 1093–1106.
- Thompson, J.S.; Schneider, P.; Kalled, S.L.; Wang, L.; Lefevre, E.A.; Cachero, T.G.; Mackay, F.; Bixler, S.A.; Zafari, M.; Liu, Z.Y.; et al. BAFF binds to the tumor necrosis factor receptor-like molecule B cell maturation antigen and is important for maintaining the peripheral B cell population. J. Exp. Med. 2000, 192, 129–135.
- Kinneer, K.; Meekin, J.; Tiberghien, A.C.; Tai, Y.-T.; Phipps, S.; Kiefer, C.M.; Rebelatto, M.C.; Dimasi, N.; Moriarty, A.; Papadopoulos, K.P.; et al. SLC46A3 as a potential predictive biomarker for antibody-drug conjugates bearing noncleavable linked maytansinoid and pyrrolobenzodiazepine warheads. Clin. Cancer Res. 2018, 24, 6570–6582.
- Hamblett, K.J.; Jacob, A.P.; Gurgel, J.L.; Tometsko, M.E.; Rock, B.M.; Patel, S.K.; Milburn, R.R.; Siu, S.; Ragan, S.P.; Rock, D.A.; et al. SLC46A3 is required to transport catabolites of noncleavable antibody maytansine conjugates from the lysosome to the cytoplasm. Cancer Res. 2015, 75, 5329–5340.
- Li, G.; Guo, J.; Shen, B.-Q.; Bumbaca Yadav, D.; Sliwkowski, M.X.; Crocker, L.M.; Lacap, J.A.; Lewis Phillips, G.D. Mechanisms of acquired resistance to trastuzumab emtansine in breast cancer cells. Mol. Cancer Ther. 2018, 17, 1441–1453.
- Lyu, M.-A.; Cheung, L.H.; Hittelman, W.N.; Marks, J.W.; Aguiar, R.C.T.; Rosenblum, M.G. The rGel/BLyS fusion toxin specifically targets malignant B cells expressing the BLyS receptors BAFF-R, TACI, and BCMA. Mol. Cancer Ther. 2007, 6, 460–470.
- Luster, T.A.; Mukherjee, I.; Carrell, J.A.; Cho, Y.H.; Gill, J.; Kelly, L.; Garcia, A.; Ward, C.; Oh, L.; Ullrich, S.J.; et al. Fusion toxin BLyS-gelonin inhibits growth of malignant human B cell lines in vitro and in vivo. PLoS ONE 2012, 7, e47361.
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209.
- Iqbal, N.; Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implications. Mol. Biol. Int. 2014, 2014, 852748.
- Marone, R.; Hess, D.; Dankort, D.; Muller, W.J.; Hynes, N.E.; Badache, A. Memo mediates ErbB2-driven cell motility. Nat. Cell Biol. 2004, 6, 515–522.
- Liu, L.; Shao, X.; Gao, W.; Bai, J.; Wang, R.; Huang, P.; Yin, Y.; Liu, P.; Shu, Y. The role of human epidermal growth factor receptor 2 as a prognostic factor in lung cancer: A meta-analysis of published data. J. Thorac. Oncol. 2010, 5, 1922–1932.
- Hommelgaard, A.M.; Lerdrup, M.; van Deurs, B. Association with membrane protrusions makes ErbB2 an internalization-resistant receptor. Mol. Biol. Cell 2004, 15, 1557–1567.
- Pust, S.; Klokk, T.I.; Musa, N.; Jenstad, M.; Risberg, B.; Erickstein, B.; Tcatchoff, L.; Liestol, K.; Danielson, H.E.; van Deurs, B.; et al. Flotillins as regulators of ErbB2 levels in breast cancer. Oncogene 2013, 32, 3443–3451.
- Cortese, K.; Howes, M.T.; Lundmark, R.; Tagliatti, E.; Bagnato, P.; Petrelli, A.; Bono, M.; McMahon, H.T.; Parton, R.G.; Tacchetti, C. The HSP90 inhibitor geldanamycin perturbs endosomal structure and drives recycling ErbB2 and transferrin to modified MVBs/lysosomal compartments. Mol. Biol. Cell 2013, 24, 129–144.
- Haslekas, C.; Breen, K.; Pedersen, K.W.; Johannessen, L.E.; Stang, E.; Madshus, I.H. The inhibitory effect of ErbB2 on epidermal growth factor-induced formation of clathrin-coated pits correlates with retention of epidermal growth factor receptor-ErbB2 oligomeric complexes at the plasma membrane. Mol. Biol. Cell 2005, 16, 5832–5842.
- Pedersen, N.M.; Madshus, I.H.; Haslekas, C.; Stang, E. Geldanamycin-induced down-regulation of ErbB2 from the plasma membrane is clathrin dependent but proteosomal activity independent. Mol. Cancer Res. 2008, 6, 491–500.
- Wang, Z.; Zhang, L.; Yeung, T.K.; Chen, X. Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol. Biol. Cell 1999, 10, 1621–1636.
- Lerdrup, M.; Bruun, S.; Grandal, M.V.; Roepstorff, K.; Kristensen, M.M.; Hommelgaard, A.M.; van Deurs, B. Endocytic down-regulation of ErbB2 is stimulated by cleavage of its C-terminus. Mol. Biol. Cell 2007, 18, 3656–3666.
- Sorkin, A.; Di Fiore, P.P.; Carpenter, G. The carboxyl terminus of epidermal growth factor receptor/erbB-2 chimerae is internalization impaired. Oncogene 1993, 8, 3021–3028.
- Lerdrup, M.; Hommelgaard, A.M.; Grandal, M.; van Deurs, B. Geldanamycin stimulates internalization of ErbB2 in a proteasome-dependent way. J. Cell Sci. 2006, 119, 85–95.
- Baulida, J.; Carpenter, G. Heregulin degradation in the absence of rapid receptor-mediated internalization. Exp. Cell Res. 1997, 232, 167–172.
- Hendriks, B.S.; Opresko, L.K.; Wiley, H.S.; Lauffenburger, D. Coregulation of epidermal growth factor receptor/human epidermal growth factor receptor 2 (HER2) levels and locations: Quantitative analysis of HER2 overexpression effects. Cancer Res. 2003, 63, 1130–1137.
- Austin, C.D.; De Maziere, A.M.; Pisacane, P.I.; van Dijk, S.M.; Eigenbrot, C.; Sliwkowski, M.X.; Klumperman, J.; Scheller, R.H. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol. Biol. Cell 2004, 15, 5268–5282.
- Karunagaran, D.; Tzahar, E.; Beerli, R.R.; Chen, X.; Graus-Porta, D.; Ratzkin, B.J.; Seger, R.; Hynes, N.E.; Yarden, Y. ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: Implications for breast cancer. EMBO J. 1996, 15, 254–264.
- Bertelsen, V.; Stang, E. The mysterious ways of ErbB2/HER2 trafficking. Membranes 2014, 4, 424–446.
- Gilboa, L.; Ben-Levy, R.; Yarden, Y.; Henis, Y.I. Roles for a cytoplasmic tyrosine and tyrosine kinase activity in the interactions of neu receptors with coated pits. J. Biol. Chem. 1995, 270, 7061–7067.
- Garay, C.; Judge, G.; Lucarelli, S.; Bautista, S.; Pandey, R.; Singh, T.; Antonescu, C.N. Epidermal growth factor-stimulated Akt phosphorylation requires clathrin or ErbB2 but not receptor endocytosis. Mol. Biol. Cell 2015, 26, 3504–3519.
- Zhao, Y.-Y.; Feron, O.; Dessy, C.; Han, X.; Marchionni, M.A.; Kelly, R.A. Neuregulin signaling in the heart. Dynamic targeting of erbB4 to caveolar microdomains in cardiac myocytes. Circ. Res. 1999, 84, 1380–1387.
- Zhou, W.; Carpenter, G. Heregulin-dependent translocation and hyperphosphorylation of ErbB-2. Oncogene 2001, 20, 3918–3920.
- Mineo, C.; Gill, G.N.; Anderson, R.G.W. Regulated migration of epidermal growth factor receptor from caveolae. J. Biol. Chem. 1999, 274, 30636–30643.
- Nagy, P.; Vereb, G.; Sebestyen, Z.; Horvath, G.; Lockett, S.J.; Damjanovich, S.; Park, J.W.; Jovin, T.M.; Szollosi, J. Lipid rafts and the local density of ErbB proteins influence the biological role of homo- and heteroassociations of ErbB2. J. Cell Sci. 2002, 115, 4251–4262.
- Harder, T.; Scheiffele, P.; Verkade, P.; Simons, K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 1998, 141, 929–942.
- Pereira, P.M.R.; Sharma, S.K.; Carter, L.M.; Edwards, K.J.; Pourat, J.; Ragupathi, A.; Janjigian, Y.Y.; Durack, J.C.; Lewis, J.S. Caveolin-1 mediates cellular distribution of HER2 and affects trastuzumab binding and therapeutic efficacy. Nat. Commun. 2018, 9, 5137.
- Barr, D.J.; Ostermeyer-Fay, A.G.; Matundan, R.A.; Brown, D.A. Clathrin-independent endocytosis of ErbB2 in geldanamycin-treated human breast cancer cells. J. Cell Sci. 2008, 121, 3155–3166.
- Wang, L.H.; Rothberg, K.G.; Anderson, R.G. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 1993, 123, 1107–1117.
- Conner, S.D.; Schmid, S.L. Differential requirements for AP-2 in clathrin-mediated endocytosis. J. Cell Biol. 2013, 162, 773–779.
- Doherty, G.J.; Lundmark, R. GRAF1-dependent endocytosis. Biochem. Soc. Trans. 2009, 37, 1061–1065.
- Burris, H.A., III; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.K.; Michaelson, R.A.; Girish, S.; et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J. Clin. Oncol. 2011, 29, 398–405.
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin. Cancer Res. 2011, 17, 6437–6447.
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.; Bezabeh, B.Z.; Leming, R.L.; Kimasi, N.; et al. A biparatopic HER2-targeting antibody-drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2-targeted therapy. Cancer Cell 2016, 29, 117–129.
- Cheng, J.; Liang, M.; Carvalho, M.F.; Tigue, N.; Faggioni, R.; Roskos, L.K.; Vainshtein, I. Molecular mechanism of HER2 rapid internalization and redirected trafficking induced by anti-HER2 biparatopic antibody. Antibodies 2020, 9, 49.
- Nami, B.; Maadi, H.; Wang, Z. Mechanisms underlying the action and synergism of trastuzumab and pertuzumab in targeting HER2-positive breast cancer. Cancers 2018, 10, 342.
- Kang, J.C.; Sun, W.; Khare, P.; Karimi, M.; Wang, X.; Shen, Y.; Ober, R.J.; Ward, E.S. Engineering a HER2-specific antibody-drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nat. Biotechnol. 2019, 37, 523–526.
- Heath, E.I.; Rosenberg, J.E. The biology and rationale of targeting nectin-4 in urothelial carcinoma. Nat. Rev. Urol. 2021, 18, 93–103.
- Franke, W.W. Discovering the molecular components of intercellular junctions—A historical review. Cold Spring Harb. Perspect. Biol. 2009, 1, a003061.
- Reymond, N.; Fabre, S.; Lecocq, E.; Adelaide, J.; Dubreuil, P.; Lopez, M. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/PRR1 through V domain interaction. J. Biol. Chem. 2001, 276, 43205–43215.
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Ratay Lortie, D.; et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 2016, 76, 3003–3013.
- Fabre-Lafay, S.; Monville, F.; Garrido-Urbani, S.; Berruyer-Pouyet, C.; Ginestier, C.; Reymond, N.; Finetti, P.; Sauvan, R.; Adelaide, J.; Geneix, J.; et al. Nectin-4 is a new histological and serological tumor associated marker for breast cancer. BMC Cancer 2007, 7, 73.
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.-T.; Sisson, G.; Tsao, M.-S.; Richardson, C.D. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011, 7, e1002240.
- Selpeut, S.; Sisson, G.; Black, K.M.; Richardson, D. Measles virus enters breast and colon cancer cell lines through a PVRL4-mediated macropinocytosis pathway. J. Virol. 2017, 91, e02191-16.

