Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francisco Muñoz | + 1431 word(s) | 1431 | 2021-07-12 12:31:16 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Muñoz, F. Mitostasis. Encyclopedia. Available online: https://encyclopedia.pub/entry/12195 (accessed on 25 June 2026).
Muñoz F. Mitostasis. Encyclopedia. Available at: https://encyclopedia.pub/entry/12195. Accessed June 25, 2026.
Muñoz, Francisco. "Mitostasis" Encyclopedia, https://encyclopedia.pub/entry/12195 (accessed June 25, 2026).
Muñoz, F. (2021, July 19). Mitostasis. In Encyclopedia. https://encyclopedia.pub/entry/12195
Muñoz, Francisco. "Mitostasis." Encyclopedia. Web. 19 July, 2021.
Copy Citation
Mitostasis refers the mitochondrial dynamics of fusion and fission depending on the cell requirements. Mitostasis also involves mitochondrial traficking and anchoring as need to maintain a functional pool of mitochondria.
mitochondria
mitostasis
calcium
oxidative stress
nitric oxide
aging
1. Introduction
The maintenance and distribution of the mitochondrial reserve is termed mitostasis, which includes mitochondrial transport, anchoring, fission and fusion. In fact, most of the mitochondria do not exist as small, distinct organelles, but rather form a highly interconnected reticulum with contiguous membranes [1]. This reticulum is highly dynamic with ongoing divisions and reconnections. The balance of fission and fusion determines the length of mitochondria, and it is regulated by stress and nutrient availability [2]. Mitostasis also involves protein reparation or degradation, and the formation of mitochondria-derived vesicles yielding to mitophagy and macroautophagy [3].
In particular, mitochondrial biogenesis requires materials from the cytoplasm and nucleus, since mammalian mitochondria DNA (mtDNA) encodes only 13 proteins [4]. For this reason, mitochondrial biogenesis requires the importation of over 1500 proteins encoded by the nuclear genome [5] and phospholipids for the inner mitochondrial membrane (IMM) and the OMM [6].
2. Mitostasis in Neurons
2.1. Mitochondrial Trafficking
The location of mitochondria, depending on local energy demands, is critical for polarized cells such as neurons. Interconnected mitochondria are located in neuronal cell soma. However, to enter the axon, a mitochondrion must undergo a fission reaction. This reaction frees it from its mitochondrial reticulum.
Once released, mitochondrial fast anterograde transport is mediated by microtubules [7]. Mitochondrial Rho GTPase 1 and 2 (Miro 1 and 2) serve as Ca2+ sensors that regulate kinesin-mediated mitochondrial motility. Miro 1 and 2 have an outer C-terminal transmembrane domain with GTPase domains, which interact with the trafficking kinesin protein (TRAK)/Milton family of proteins [8][9][10][11][12]. These cargo adaptorsbind to Kinesin-1 anddynein/dynactin, beingcritical in mitochondrial trafficking and its spatial distribution [13]. Kinesin-1 cargoes, often together with dynein, drive mitochondria transport into the axon [11][14], and the dynein/dynactin cargo complextransports mitochondria into dendrites [13][15].
2.2. Mitochondrial Anchoring
Mitochondrial anchoring to inner cell structures is needed to provide the required energy at the right place, as around two-thirds of neuronal mitochondria remain stationary [18]. These pools of mitochondria are mainly located in the soma and in areas with high ATP demand, such as the nodes of Ranvier, axonal presynaptic endings and dendrites [19]. Mitochondria anchoring mechanisms are not fully understood and they seem to involve different proteins depending on the localization of the mitochondria. For instance, in axonal presynaptic endings, activation of adenosine monophosphate-activated protein kinase (AMPK) by synaptic activity leads to myosin VI phosphorylation, ending with mitochondrial recruitment and syntaphilin-mediated anchoring on presynaptic filamentous actin [20]. This process is specific to the stationary mitochondria, not being related to the mitochondria mobility machinery [21][22]. Remarkably, it is an axon exclusive physiological mechanism. On the other hand, axonal mitochondrial anchor mislocalization to dendrites has been associated with multiple sclerosis disease [23].
2.3. Mitochondrial Fission
Mitochondrial fission is essential for shaping the mitochondrial network and is required for generating additional mitochondria of the appropriate size for their transport by molecular motors along the cytoskeleton [24] (Figure 1).
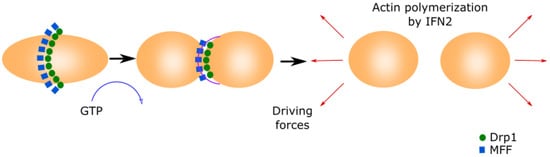
Figure 1. Mitochondrial fission.This process is initiated by MFF forming a ring in the middle of the primary organelle to be divided. Drp1 GTPase translocates to the mitochondria and binds to MFF, inducing a conformational change by the hydrolysis of GTP. This conformational change generates the ring contraction, which starts mitochondrial fission. The reorganization of the cytoskeleton generates the driving forces that separate both new mitochondria.
Mitochondria fission starts when the mitochondrial fission factor (MFF) surrounds the OMM forming a ring [25][26]. Mitochondrial fission 1 (Fis1) and mitochondrial dynamics proteins of 49 and 51 kDa (MID49 and MID51) recruit the cytoplasmic Dynamin-related protein 1 (Drp1) [27][28][29]. Drp1 translocates to the mitochondria and binds to MFF, inducing a conformational change due to its GTPase activity. This conformational change produces the ring contraction that starts mitochondrial fission [27][30]. Cellular cytoskeleton further separates both mitochondria depending on the MAMs. The Inverted Formin 2 protein (INF2) is localized in MAMs and its activation causes actin polymerization, which generates the driven force separating both mitochondria [31].
2.4. Mitochondrial Fusion
Mitochondrial fusion is a cell protective mechanism [32] based in two different and concomitant processes, the OMM fusion and the IMM fusion (Figure 2).
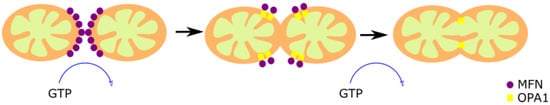
Figure 2. Mitochondrial fusion. The fusion of two preexistent mitochondria is a protective mechanism based on the fusion of the OMM and IMM. The OMM fusion is mediated by MFN1 and MFN2, and the fusion of the IMM is mediated by OPA1. First, MFN1 and MFN2 GTPase activity induce the formation of homo- or hetero-oligomeric complexes in the fusion spot. Then, OPA1-mediated GTP hydrolysis promotes the membrane fusion.
The dynamin-associated GTPases Mitofusin proteins 1 and 2 (MFN1 and MFN2) are responsible for the OMM fusion [33][34]. Both GTPases share a high structural and biochemical homology, but they have different roles [35]. MFN1 is found exclusively in the OMM, and allows the contact between the OMM of different mitochondria [36]. MFN2, found in mitochondria and ER, has an anchoring role since it joins the two organelles, forming homotypic and heterotypic complexes with MFN1 [37]. The spiral domains of MFN1 and MFN2 interact, forming homo- or hetero-oligomeric complexes through GTP hydrolysis in the fusion spot [38]. However, there are controversial reports regarding the relevance of this mechanism since it was observed that the loss of MFN-2 increased the ER–mitochondrial juxtaposition [39]. This fact does not contradict that the functionality of mitochondria–ER contacts is affected in MFN2 KO cells, as suggested by the previous observation that Ca2+ transfer from the ER to the mitochondria is altered in these cells [37].
The optic atrophy 1 protein (OPA1) mediates the IMM fusion [40][41]. The long form of the protein (l-OPA1) works as a coordinator of the IMM of both fusing mitochondria. The two membranes fuse through the short isoform (s-OPA1) by its GTPase activity [42]. OPA1 is also associated with the maintenance of the MRC by cristae organization, and the control of apoptosis [43][44][45][46], demonstrating versatility of functions.
2.5. The regulator of Mitochondrial Biogenesis PGC-1α
The peroxisome proliferator-activated receptor-γ (PPARγ) coactivator-1 α (PGC-1α) is a member of a transcription coactivator family, composed ofPGC-1α, PGC-1β and PGC-related coactivator (PRC). These proteins play a central role in the regulation of mitochondrial biogenesis and cellular energy metabolism [47] (Figure 3).
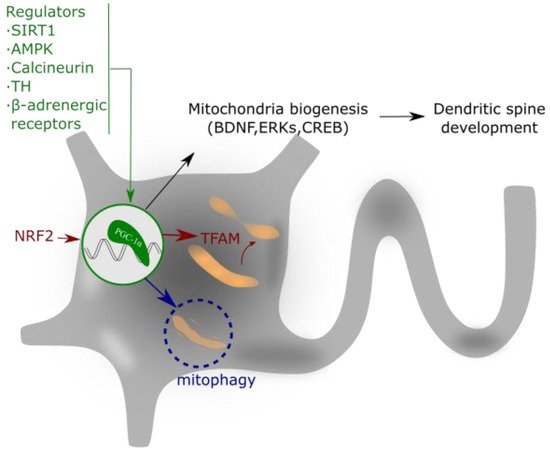
Figure 3. PGC-1α regulates mitochondrial biogenesis and spine growth. Its activity is based on the interaction with NRF that activates to TFAM. This complex regulates mitochondrial gene expression and addresses the requirements of neuronal energy when needed for dendrite activity or growth. PGC-1α expression is regulated by different intracellular signaling pathways, as explained in the text.
The action mechanism of PGC-1α implies its interaction with two nuclear transcription factors, nuclear respiratory factors 1 and 2 (NRF-1 and 2), which activate the mitochondrial transcription factor A (TFAM), a mtDNA-binding protein [48]. TFAM and the cofactor mitochondrial transcription factor B (TFB1M) are essential for mitochondrial gene expression [49]. This molecular pathway promotes the equilibrium in the assembly of some components of the MRC, which ultimately ensures appropriate mitochondrial function, mtDNA replication and apoptosis control [50][51][52].
The expression and/or activity of PGC-1α is regulated by different intracellular signaling pathways such as the PGC-1α regulatory cascade [53], thyroid hormone (TH) [54], sirtuin 1 (SIRT1) [55][56], calcineurin [57], AMPK [58], cyclin-dependent kinases (CDKs) [59], and β-adrenergic signaling [60]. These regulators of PGC-1α aim to coordinate the expression and function of mitochondrial proteins during mitochondrial biogenesis [61]. Additionally, not only transcriptional regulation but also post-translational modifications allow the control of the PGC-1α activity. For example, PGC-1α phosphorylation by p38 mitogen-activated protein kinase alpha (p38α) enhances its activity [62], and PGC-1α acetylation suppresses its activity [55].
PGC-1α and mitochondrial biogenesis play important roles in the formation and maintenance of dendritic spines and synapses in hippocampal neurons [63]. In this context, PGC-1α also controls the synthesis of the Brain-Derived Neurotrophic Factor (BDNF) [64] and the activity of Extracellular Signal-Regulated Kinases (ERKs) [65]. BNDF has been widely reported in the literature as a neuroprotective agent produced by physical exercise [66] and, interestingly, exercise induces the upregulation of PGC-1α expression and increases mitochondrial mass within the brain [67]. In addition to its role in mitochondrial biogenesis, PGC-1α also regulates apoptosis and the ROS detoxification system [68].
3. Conclusions
Aging and neurodegenerative processes are not the consequences of a unique signaling pathway alteration but of multifactorial events, including dysfunction of mitochondria that yield to failures in ATP production, increased ROS production and alterations in Ca2+ homeostasis. Considering mitochondria as a therapeutic target would produce an improvement in the maintenance of the optimum neuronal function. These therapeutic approaches would include the regulation of PGC-1α and calcium buffering systems as well as free radicals’generation, addressing the source of free radicals more than the use of antioxidants that have not demonstrated effective results to date.
References
- Braschi, E.; McBride, H.M. Mitochondria and the culture of the Borg. BioEssays 2010, 32, 958–966.
- Wai, T.; Langer, T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016, 27, 105–117.
- Misgeld, T.; Schwarz, T.L. Mitostasis in neurons: Maintaining mitochondria in an extended cellular architecture. Neuron 2017, 96, 651–666.
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 2010, 11, 25–44.
- Nicolas, E.; Tricarico, R.; Savage, M.; Golemis, E.A.; Hall, M.J. Disease-associated genetic variation in human mitochondrial protein import. Am. J. Hum. Genet. 2019, 104, 784–801.
- Connerth, M.; Tatsuta, T.; Haag, M.; Klecker, T.; Westermann, B.; Langer, T. Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science (80-. ). 2012, 338, 815–818.
- Vicario-Orri, E.; Opazo, C.M.; Muñoz, F.J. The pathophysiology of axonal transport in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 43, 1097–1113.
- Guo, X.; Macleod, G.T.; Wellington, A.; Hu, F.; Panchumarthi, S.; Schoenfield, M.; Marin, L.; Charlton, M.P.; Atwood, H.L.; Zinsmaier, K.E. The GTPase dMiro is required for axonal transport of mitochondria to drosophila synapses. Neuron 2005, 47, 379–393.
- Brickley, K.; Stephenson, F.A. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J. Biol. Chem. 2011, 286, 18079–18092.
- Stowers, R.S.; Megeath, L.J.; Górska-Andrzejak, J.; Meinertzhagen, I.A.; Schwarz, T.L. Axonal transport of mitochondria to synapses depends on Milton, a novel Drosophila protein. Neuron 2002, 36, 1063–1077.
- Nangaku, M.; Sato-Yoshitake, R.; Okada, Y.; Noda, Y.; Takemura, R.; Yamazaki, H.; Hirokawa, N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 1994, 79, 1209–1220.
- Fransson, Å.; Ruusala, A.; Aspenström, P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 2006, 344, 500–510.
- Van Spronsen, M.; Mikhaylova, M.; Lipka, J.; Schlager, M.A.; van den Heuvel, D.J.; Kuijpers, M.; Wulf, P.S.; Keijzer, N.; Demmers, J.; Kapitein, L.C.; et al. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 2013, 77, 485–502.
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 2006, 17, 2057–2068.
- Kapitein, L.C.; Schlager, M.A.; Kuijpers, M.; Wulf, P.S.; van Spronsen, M.; MacKintosh, F.C.; Hoogenraad, C.C. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr. Biol. 2010, 20, 290–299.
- Quintero, O.A.; DiVito, M.M.; Adikes, R.C.; Kortan, M.B.; Case, L.B.; Lier, A.J.; Panaretos, N.S.; Slater, S.Q.; Rengarajan, M.; Feliu, M.; et al. Human Myo19 is a novel myosin that associates with mitochondria. Curr. Biol. 2009, 19, 2008–2013.
- López-Doménech, G.; Covill-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336.
- Morris, R.L.; Hollenbeck, P.J. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 1993, 104, 917–927.
- Kraft, L.M.; Lackner, L.L. Mitochondrial anchors: Positioning mitochondria and more. Biochem. Biophys. Res. Commun. 2018, 500, 2–8.
- Li, S.; Xiong, G.J.; Huang, N.; Sheng, Z.H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020, 2, 1077–1095.
- Lin, M.Y.; Sheng, Z.H. Regulation of mitochondrial transport in neurons. Exp. Cell Res. 2015, 334, 35–44.
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 2013, 5.
- Joshi, D.C.; Zhang, C.L.; Babujee, L.; Vevea, J.D.; August, B.K.; Sheng, Z.H.; Chapman, E.R.; Gomez, T.M.; Chiu, S.Y. Inappropriate intrusion of an axonal mitochondrial anchor into dendrites causes neurodegeneration. Cell Rep. 2019, 29, 685–696.e5.
- Fukumitsu, K.; Hatsukano, T.; Yoshimura, A.; Heuser, J.; Fujishima, K.; Kengaku, M. Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol. Cell. Neurosci. 2016, 71, 56–65.
- Gandre-Babbe, S.; Van Der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412.
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158.
- Smirnova, E.; Griparic, L.; Shurland, D.L.; Van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256.
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 2003, 23, 5409–5420.
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573.
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362.
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467.
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562.
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874.
- Hales, K.G.; Fuller, M.T. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell 1997, 90, 121–129.
- Zorzano, A.; Pich, S. What is the biological significance of the two mitofusin proteins present in the outer mitochondrial membrane of mammalian cells? IUBMB Life 2006, 58, 441–443.
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546.
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610.
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862.
- Cosson, P.; Marchetti, A.; Ravazzola, M.; Orci, L. Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: An ultrastructural study. PLoS One 2012, 7.
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002, 523, 171–176.
- Meeusen, S.; DeVay, R.; Block, J.; Cassidy-Stone, A.; Wayson, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 2006, 127, 383–395.
- DeVay, R.M.; Dominguez-Ramirez, L.; Lackner, L.L.; Hoppins, S.; Stahlberg, H.; Nunnari, J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 2009, 186, 793–803.
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtdna stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289.
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691.
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171.
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746.
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124.
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313.
- McCulloch, V.; Shadel, G.S. Human mitochondrial transcription factor B1 interacts with the C-terminal activation region of h-mtTFA and stimulates transcription independently of Its RNA methyltransferase activity. Mol. Cell. Biol. 2003, 23, 5816–5824.
- Guo, J.; Zheng, L.; Liu, W.; Wang, X.; Wang, Z.; Wang, Z.; French, A.J.; Kang, D.; Chen, L.; Thibodeau, S.N.; et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res. 2011, 71, 2978–2987.
- Kanki, T.; Ohgaki, K.; Gaspari, M.; Gustafsson, C.M.; Fukuoh, A.; Sasaki, N.; Hamasaki, N.; Kang, D. Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA. Mol. Cell. Biol. 2004, 24, 9823–9834.
- Kaufman, B.A.; Durisic, N.; Mativetsky, J.M.; Costantino, S.; Hancock, M.A.; Grutter, P.; Shoubridge, E.A. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol. Biol. Cell 2007, 18, 3225–3236.
- Schilling, J.; Kelly, D.P. The PGC-1 cascade as a therapeutic target for heart failure. J. Mol. Cell. Cardiol. 2011, 51, 578–583.
- McClure, T.D.; Young, M.E.; Taegtmeyer, H.; Ning, X.-H.; Buroker, N.E.; López-Guisa, J.; Portman, M.A. Thyroid hormone interacts with PPARα and PGC-1 during mitochondrial maturation in sheep heart. Am. J. Physiol. Circ. Physiol. 2005, 289, H2258–H2264.
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118.
- Gurd, B.J.; Yoshida, Y.; McFarlan, J.T.; Holloway, G.P.; Moyes, C.D.; Heigenhauser, G.J.F.; Spriet, L.; Bonen, A. Nuclear SIRT1 activity, but not protein content, regulates mitochondrial biogenesis in rat and human skeletal muscle. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 2011, 301.
- Schaeffer, P.J.; Wende, A.R.; Magee, C.J.; Neilson, J.R.; Leone, T.C.; Chen, F.; Kelly, D.P. Calcineurin and calcium/calmodulin-dependent protein kinase activate distinct metabolic gene regulatory programs in cardiac muscle. J. Biol. Chem. 2004, 279, 39593–39603.
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987.
- Sano, M.; Wang, S.C.; Shirai, M.; Scaglia, F.; Xie, M.; Sakai, S.; Tanaka, T.; Kulkarni, P.A.; Barger, P.M.; Youker, K.A.; et al. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 2004, 23, 3559–3569.
- Brandt, N.; Nielsen, L.; Thiellesen Buch, B.; Gudiksen, A.; Ringholm, S.; Hellsten, Y.; Bangsbo, J.; Pilegaard, H. Impact of β-adrenergic signaling in PGC-1α-mediated adaptations in mouse skeletal muscle. Am. J. Physiol. Metab. 2018, 314, E1–E20.
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol. Cell 2001, 8, 971–982.
- Fan, M.; Rhee, J.; St-Pierre, J.; Handschin, C.; Puigserver, P.; Lin, J.; Jäeger, S.; Erdjument-Bromage, H.; Tempst, P.; Spiegelman, B.M. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1α: Modulation by p38 MAPK. Genes Dev. 2004, 18, 278–289.
- Cheng, A.; Wan, R.; Yang, J.L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3.
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013, 18, 649–659.
- Zhang, Y.; Li, R.; Meng, Y.; Li, S.; Donelan, W.; Zhao, Y.; Qi, L.; Zhang, M.; Wang, X.; Cui, T.; et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 2014, 63, 514–525.
- Pedersen, B.K. Physical activity and muscle–brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392.
- Steiner, J.L.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. Exercise training increases mitochondrial biogenesis in the brain. J. Appl. Physiol. 2011, 111, 1066–1071.
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408.
More
Information
Subjects:
Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revision:
1 time
(View History)
Update Date:
19 Jul 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No