 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hadia Moindjie | + 2371 word(s) | 2371 | 2021-07-05 08:48:25 | | | |
| 2 | Vivi Li | + 75 word(s) | 2446 | 2021-07-16 06:09:20 | | |
Video Upload Options
Carcinogenesis is a multi-step process that refers to transformation of a normal cell into a tumoral neoplastic cell. The mechanisms that promote tumor initiation, promotion and progression are varied, complex and remain to be understood. Studies have highlighted the involvement of oncogenic mutations, genomic instability and epigenetic alterations as well as metabolic reprogramming, in different processes of oncogenesis. However, the underlying mechanisms still have to be clarified. Mitochondria are central organelles at the crossroad of various energetic metabolisms. In addition to their pivotal roles in bioenergetic metabolism, they control redox homeostasis, biosynthesis of macromolecules and apoptotic signals, all of which are linked to carcinogenesis.
1. Introduction
2. Mutations in Genes Involved in Mitochondrial Metabolism Drive Carcinogenesis Initiation
2.1. Mutations and Decreased Copy Number of mtDNA
2.2. Mutations in Nuclear-Encoded Mitochondrial Genes
2.2.1. Bioenergetic Metabolism Alteration
2.2.2. Oxidative Stress Promotion
2.2.3. Epigenetic Regulation
3. Mitochondria as Promising Targets in Cancer Therapies
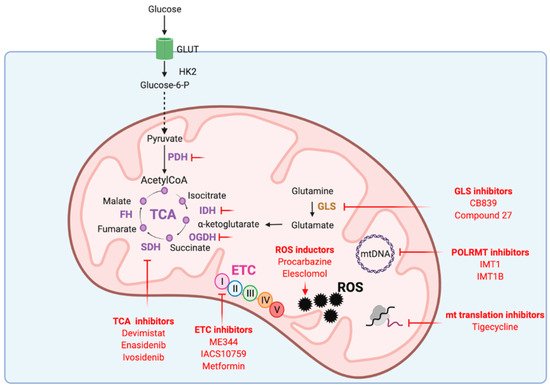
3.1. Targeting mtDNA Transcription and Translation
3.2. Targeting ETC
3.3. Targeting the TCA Cycle
3.4. Targeting Redox Homeostasis
References
- Pitot, H.C. The Molecular Biology of Carcinogenesis. Cancer 1993, 72, 962–970.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Rodrigues-Ferreira, S.; Moindjie, H.; Haykal, M.M.; Nahmias, C. Predicting and Overcoming Taxane Chemoresistance. Trends Mol. Med. 2021, 27, 138–151.
- Zhou, Z.; Ibekwe, E.; Chornenkyy, Y. Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change. Antioxidants 2018, 7, 16.
- Corbet, C.; Feron, O. Cancer Cell Metabolism and Mitochondria: Nutrient Plasticity for TCA Cycle Fueling. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 7–15.
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754.
- Newmeyer, D.D.; Ferguson-Miller, S. Mitochondria: Releasing Power for Life and Unleashing the Machineries of Death. Cell 2003, 112, 481–490.
- Chandra, D.; Liu, J.-W.; Tang, D.G. Early Mitochondrial Activation and Cytochrome c Up-Regulation during Apoptosis*210. J. Biol. Chem. 2002, 277, 50842–50854.
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA Copy Number Variation across Human Cancers. eLife 2016, 5, e10769.
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. MtDNA Mutations Increase Tumorigenicity in Prostate Cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724.
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The Maintenance of Mitochondrial DNA Integrity—Critical Analysis and Update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641.
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA Mutations in Human Cancer. Oncogene 2006, 25, 4663–4674.
- Warowicka, A.; Wołuń-Cholewa, M.; Kwaśniewska, A.; Goździcka-Józefiak, A. Alternations in Mitochondrial Genome in Carcinogenesis of HPV Positive Cervix. Exp. Mol. Pathol. 2020, 117, 104530.
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A Heteroplasmic, Not Homoplasmic, Mitochondrial DNA Mutation Promotes Tumorigenesis via Alteration in Reactive Oxygen Species Generation and Apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589.
- Dasgupta, S.; Hoque, M.O.; Upadhyay, S.; Sidransky, D. Mitochondrial Cytochrome B Gene Mutation Promotes Tumor Growth in Bladder Cancer. Cancer Res. 2008, 68, 700–706.
- Hertweck, K.L.; Dasgupta, S. The Landscape of MtDNA Modifications in Cancer: A Tale of Two Cities. Front. Oncol. 2017, 7.
- Kopinski, P.K.; Janssen, K.A.; Schaefer, P.M.; Trefely, S.; Perry, C.E.; Potluri, P.; Tintos-Hernandez, J.A.; Singh, L.N.; Karch, K.R.; Campbell, S.L.; et al. Regulation of Nuclear Epigenome by Mitochondrial DNA Heteroplasmy. Proc. Natl. Acad. Sci. USA 2019, 116, 16028–16035.
- Smiraglia, D.J.; Kulawiec, M.; Bistulfi, G.L.; Gupta, S.G.; Singh, K.K. A Novel Role for Mitochondria in Regulating Epigenetic Modification in the Nucleus. Cancer Biol. 2008, 7, 1182–1190.
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH Mutation in Glioma: Molecular Mechanisms and Potential Therapeutic Targets. Br. J. Cancer 2020, 122, 1580–1589.
- Bardella, C.; Pollard, P.J.; Tomlinson, I. SDH Mutations in Cancer. Biochim. Biophys. Acta 2011, 1807, 1432–1443.
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem Oxygenase Is Synthetically Lethal with the Tumour Suppressor Fumarate Hydratase. Nature 2011, 477, 225–228.
- Tseng, P.-L.; Wu, W.-H.; Hu, T.-H.; Chen, C.-W.; Cheng, H.-C.; Li, C.-F.; Tsai, W.-H.; Tsai, H.-J.; Hsieh, M.-C.; Chuang, J.-H.; et al. Decreased Succinate Dehydrogenase B in Human Hepatocellular Carcinoma Accelerates Tumor Malignancy by Inducing the Warburg Effect. Sci. Rep. 2018, 8.
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-Alpha Prolyl Hydroxylase. Cancer Cell 2005, 7, 77–85.
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.-L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF Overexpression Correlates with Biallelic Loss of Fumarate Hydratase in Renal Cancer: Novel Role of Fumarate in Regulation of HIF Stability. Cancer Cell 2005, 8, 143–153.
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25.
- Ishii, T.; Yasuda, K.; Akatsuka, A.; Hino, O.; Hartman, P.S.; Ishii, N. A Mutation in the SDHC Gene of Complex II Increases Oxidative Stress, Resulting in Apoptosis and Tumorigenesis. Cancer Res. 2005, 65, 203–209.
- Purohit, V.; Simeone, D.M.; Lyssiotis, C.A. Metabolic Regulation of Redox Balance in Cancer. Cancers 2019, 11, 955.
- Zheng, L.; Cardaci, S.; Jerby, L.; MacKenzie, E.D.; Sciacovelli, M.; Johnson, T.I.; Gaude, E.; King, A.; Leach, J.D.G.; Edrada-Ebel, R.; et al. Fumarate Induces Redox-Dependent Senescence by Modifying Glutathione Metabolism. Nat. Commun. 2015, 6, 6001.
- Kanamori, M.; Higa, T.; Sonoda, Y.; Murakami, S.; Dodo, M.; Kitamura, H.; Taguchi, K.; Shibata, T.; Watanabe, M.; Suzuki, H.; et al. Activation of the NRF2 Pathway and Its Impact on the Prognosis of Anaplastic Glioma Patients. Neuro-Oncology 2015, 17, 555–565.
- Gilbert, M.R.; Liu, Y.; Neltner, J.; Pu, H.; Morris, A.; Sunkara, M.; Pittman, T.; Kyprianou, N.; Horbinski, C. Autophagy and Oxidative Stress in Gliomas with IDH1 Mutations. Acta Neuropathol. 2014, 127, 221–233.
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, Subunit of Complex II Triggers Reactive Oxygen Species-Dependent Hypoxia-Inducible Factor Activation and Tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731.
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs Cycle-Deficient Hereditary Cancer Syndromes Are Defined by Homologous Recombination DNA Repair Defects. Nat. Genet. 2018, 50, 1086–1092.
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.B.; et al. Fumarate Is an Epigenetic Modifier That Elicits Epithelial-to-Mesenchymal Transition. Nature 2016, 537, 544–547.
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752.
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30.
- Bonekamp, N.A.; Peter, B.; Hillen, H.S.; Felser, A.; Bergbrede, T.; Choidas, A.; Horn, M.; Unger, A.; Di Lucrezia, R.; Atanassov, I.; et al. Small-Molecule Inhibitors of Human Mitochondrial DNA Transcription. Nature 2020, 588, 712–716.
- Skrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688.
- Hu, B.; Guo, Y. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Ovarian Cancer to Overcome Chemoresistance. Biochem. Biophys. Res. Commun. 2019, 509, 373–378.
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting Mitochondrial Oxidative Phosphorylation Eradicates Therapy-Resistant Chronic Myeloid Leukemia Stem Cells. Nat. Med. 2017, 23, 1234–1240.
- Reed, G.A.; Schiller, G.J.; Kambhampati, S.; Tallman, M.S.; Douer, D.; Minden, M.D.; Yee, K.W.; Gupta, V.; Brandwein, J.; Jitkova, Y.; et al. A Phase 1 Study of Intravenous Infusions of Tigecycline in Patients with Acute Myeloid Leukemia. Cancer Med. 2016, 5, 3031–3040.
- Dong, L.; Neuzil, J. Targeting Mitochondria as an Anticancer Strategy. Cancer Commun. 2019, 39, 63.
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. Elife 2014, 3, e02242.
- Cai, H.; Everett, R.S.; Thakker, D.R. Efficacious Dose of Metformin for Breast Cancer Therapy Is Determined by Cation Transporter Expression in Tumours. Br. J. Pharm. 2019, 176, 2724–2735.
- Madera, D.; Vitale-Cross, L.; Martin, D.; Schneider, A.; Molinolo, A.A.; Gangane, N.; Carey, T.E.; McHugh, J.B.; Komarck, C.M.; Walline, H.M.; et al. Prevention of Tumor Growth Driven by PIK3CA and HPV Oncogenes by Targeting MTOR Signaling with Metformin in Oral Squamous Carcinomas Expressing OCT3. Cancer Prev. Res. 2015, 8, 197–207.
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352.
- Lim, S.C.; Carey, K.T.; McKenzie, M. Anti-Cancer Analogues ME-143 and ME-344 Exert Toxicity by Directly Inhibiting Mitochondrial NADH: Ubiquinone Oxidoreductase (Complex I). Am. J. Cancer Res. 2015, 5, 689–701.
- Ghosh, P.; Vidal, C.; Dey, S.; Zhang, L. Mitochondria Targeting as an Effective Strategy for Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 3363.
- Jeyaraju, D.V.; Hurren, R.; Wang, X.; MacLean, N.; Gronda, M.; Shamas-Din, A.; Minden, M.D.; Giaever, G.; Schimmer, A.D. A Novel Isoflavone, ME-344, Targets the Cytoskeleton in Acute Myeloid Leukemia. Oncotarget 2016, 7, 49777–49785.
- Quintela-Fandino, M.; Morales, S.; Cortés-Salgado, A.; Manso, L.; Apala, J.V.; Muñoz, M.; Gasol Cudos, A.; Salla Fortuny, J.; Gion, M.; Lopez-Alonso, A.; et al. Randomized Phase 0/I Trial of the Mitochondrial Inhibitor ME-344 or Placebo Added to Bevacizumab in Early HER2-Negative Breast Cancer. Clin. Cancer Res. 2020, 26, 35–45.
- Carter, J.L.; Hege, K.; Kalpage, H.A.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting Mitochondrial Respiration for the Treatment of Acute Myeloid Leukemia. Biochem. Pharm. 2020, 182, 114253.
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.A.; et al. An Inhibitor of Oxidative Phosphorylation Exploits Cancer Vulnerability. Nat. Med. 2018, 24, 1036–1046.
- Panina, S.B.; Pei, J.; Baran, N.; Konopleva, M.; Kirienko, N.V. Utilizing Synergistic Potential of Mitochondria-Targeting Drugs for Leukemia Therapy. Front. Oncol. 2020, 10, 435.
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A Strategically Designed Small Molecule Attacks Alpha-Ketoglutarate Dehydrogenase in Tumor Cells through a Redox Process. Cancer Metab. 2014, 2, 4.
- Pardee, T.S.; Lee, K.; Luddy, J.; Maturo, C.; Rodriguez, R.; Isom, S.; Miller, L.D.; Stadelman, K.M.; Levitan, D.; Hurd, D.; et al. A Phase I Study of the First-in-Class Antimitochondrial Metabolism Agent, CPI-613, in Patients with Advanced Hematologic Malignancies. Clin. Cancer Res. 2014, 20, 5255–5264.
- Lycan, T.W.; Pardee, T.S.; Petty, W.J.; Bonomi, M.; Alistar, A.; Lamar, Z.S.; Isom, S.; Chan, M.D.; Miller, A.A.; Ruiz, J. A Phase II Clinical Trial of CPI-613 in Patients with Relapsed or Refractory Small Cell Lung Carcinoma. PLoS ONE 2016, 11, e0164244.
- Philip, P.A.; Buyse, M.E.; Alistar, A.T.; Rocha Lima, C.M.; Luther, S.; Pardee, T.S.; Van Cutsem, E. A Phase III Open-Label Trial to Evaluate Efficacy and Safety of CPI-613 plus Modified FOLFIRINOX (MFFX) versus FOLFIRINOX (FFX) in Patients with Metastatic Adenocarcinoma of the Pancreas. Future Oncol. 2019, 15, 3189–3196.
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417.
- Lee, P.; Malik, D.; Perkons, N.; Huangyang, P.; Khare, S.; Rhoades, S.; Gong, Y.-Y.; Burrows, M.; Finan, J.M.; Nissim, I.; et al. Targeting Glutamine Metabolism Slows Soft Tissue Sarcoma Growth. Nat. Commun. 2020, 11, 498.
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat. Rev. Cancer 2016, 16, 773.
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer 2014, 13, 890–901.
- Soth, M.J.; Le, K.; Di Francesco, M.E.; Hamilton, M.M.; Liu, G.; Burke, J.P.; Carroll, C.L.; Kovacs, J.J.; Bardenhagen, J.P.; Bristow, C.A.; et al. Discovery of IPN60090, a Clinical Stage Selective Glutaminase-1 (GLS-1) Inhibitor with Excellent Pharmacokinetic and Physicochemical Properties. J. Med. Chem. 2020, 63, 12957–12977.
- Glasauer, A.; Chandel, N.S. Targeting Antioxidants for Cancer Therapy. Biochem. Pharm. 2014, 92, 90–101.
- Sborov, D.W.; Haverkos, B.M.; Harris, P.J. Investigational Cancer Drugs Targeting Cell Metabolism in Clinical Development. Expert Opin. Investig. Drugs 2015, 24, 79–94.
- Dong, L.; Gopalan, V.; Holland, O.; Neuzil, J. Mitocans Revisited: Mitochondrial Targeting as Efficient Anti-Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 7941.

