 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | James Pepper Bennett Jr | + 2159 word(s) | 2159 | 2021-05-13 09:50:50 | | | |
| 2 | Dean Liu | -10 word(s) | 2149 | 2021-07-14 04:43:28 | | |
Video Upload Options
Alzheimer’s disease (AD) is a neurodegenerative disease associated with human aging. Ten percent of individuals over 65 years have AD and its prevalence continues to rise with increasing age.
1. Introduction
Aging is characterized by dysregulated immune [1] and metabolic homeostasis [2][3] where there is chronic sterile low-grade inflammation or inflammaging [4] that involves cellular senescence [5][6], immunosenescence [7][8][9][10], mitochondrial dysfunction [11][12], defective autophagy [13][14] and mitophagy [15][16], dysregulation of the ubiquitin–proteasome system [17][18], activation of the DNA damage response [19][20], meta-inflammation or metaflammation from chronic overnutrition or obesity [21][22], and gut microbiota dysbiosis [5][23][24][25]. These are reflected by changes in circulating immune markers including C-reactive protein (CRP) [26], interleukin-6 (IL-6) [27], tumor necrosis factor alpha (TNF-α) [28] and its soluble receptors (tumor necrosis factor receptor I (TNFR-I) and tumor necrosis factor receptor II (TNFR-II)) [28], vascular cell adhesion molecule I (VCAM-I) [29], d-dimer [30], and sirtuin signaling [31][32]. The drawback of chronic subclinical inflammation is that it is an essential risk factor for increasing the incidence of degenerative diseases such as AD [33][34][35]. There are currently an estimated 728 million persons aged 65 years or over in the world. In the next 30 years, this number is expected to more than double to exceed 1.5 billion in 2050 (https://www.un.org/development/desa/pd/news/world-population-ageing-2020-highlights (accessed on 20 March 2021)). Thus, the aging population vulnerable to inflammaging will significantly increase over the next few decades.
AD is a chronic devastating neurodegenerative disorder in which increasing age is the strongest non-modifiable disease risk factor [36][37]. There are currently no effective therapies for AD [38][39]. It is clinically characterized by the progressive deterioration of memory and other cognitive functions [40]. It is the leading cause of dementia, affecting 50 million people worldwide [41]. Its neuropathological hallmarks include extracellular β-amyloid (Aβ) plaques and intracellular hyper-phosphorylated tau (p-τ) in neurofibrillary tangles, accompanied with synaptic and neuronal loss [40][41][42]. AD can be classified as (i) familial or early-onset AD (EOAD) or (ii) sporadic or late-onset AD (LOAD) [43]. Both share almost similar pathophysiology [44][45]. Three causative genes, including presenilin 1 (PSEN1) [46], presenilin 2 (PSEN2) [47], and amyloid precursor protein (APP) [48], are involved in the pathogenesis of EOAD in an autosomal-dominant trait [49][50]. LOAD, however, comprises most AD cases (>95%) where the greatest risk factor is advanced age [51], while the common genetic risk factor is an allelic variation in apolipoprotein E (Apo E) [52]. Recent large scale studies of AD genetics, employing genome-wide association studies (GWAS), whole exome sequencing (WES), and whole genome sequencing (WGS), have defined additional genes whose variants contribute to increased risk [53][54]. These include Clusterin (CLU), Sortilin-related receptor-1 (SORL1), ATP-binding cassette subfamily A member 7 (ABCA7), Bridging integrator 1 (BIN1), phosphatidylinositol binding clathrin assembly protein (PICALM), CD2 associated protein (CD2AP), Complement component (3b/4b) receptor 1 (CR1), CD33, triggering receptor expressed on myeloid cells 2 (TREM2), and phospholipase D3 (PLD3) [55][56][57]. Intriguingly, more than 50% of validated gene variants are implicated in innate immune and microglial functions [58][59][60], including the top two AD risk genes, APOE and TREM2 [61][62]. Epigenomic analysis shows that AD GWAS loci are preferentially enriched in enhancer sequences involved in innate immune processes [63][64] as well as endocytosis, cholesterol/sterol metabolism, and synaptic function [55][65][66]. TREM2 enhances the rate of phagocytosis in microglia and macrophages; modulates inflammatory signaling; and controls myeloid cell number, proliferation, and survival [67], and it has been revealed that triggering TREM2 receptor in microglial cells is closely associated with the pathogenesis of AD [68]. TREM2 modulates microglial functions in response to Aβ plaques and tau tangles [69][70]. In early AD, the absence of TREM2 leads to increased amyloid pathology that progressively becomes worse owing to the loss of phagocytic Aβ clearance [69]. In AD, TREM2 variants arise in part because of their reduced capacity to phagocytose Aβ [71].
While compelling evidence indicates that AD has a multifactorial etiology [72][73][74], neuroinflammation plays a central role in its etiopathogenesis [75][76], owing to its capacity to exacerbate Aβ and τ pathologies [77]. In vivo positron emission tomography (PET) studies provide direct evidence of increased microglia activation (inflammation) in the brains of AD patients [78][79][80]. The levels of pro-inflammatory cytokines in AD patient serum and post mortem brain are elevated [81][82], and Aβ can activate the brains’ innate immune cells [83][84]. The sustained inflammatory response in AD brains [85][86][87] extends beyond a reaction to neuronal loss [88] and involves microglia, astrocytes, oligodendrocytes, mast cells, cytokines, and chemokines, as well as complement [89]. These collectively play an integral role in the onset and progression of the disease [88][90][91]. Other early-onset processes involved in the etiology of the disease include mitochondrial dysfunction resulting in altered glucose metabolism [92] and oxidative stress [93]; chronic hypoperfusion [94]; and neuronal cell cycle re-entry that leads to neuronal tetraploidization (NT) [94], trisomy 21 mosaicism [95], and synapse loss [96]. These processes may synergistically interact to facilitate the neurodegenerative process in AD [93][97].
2. Mediators of Neuroinflammation
2.1. Microglia
Microglia are the CNS’s immune cells [98] and are different from peripheral and other tissue-resident macrophages [99][100][101][102][103][104]. They arise from yolk-sac fetal macrophages and are unique in their capacity for self-renewal [105][106][107]. Other tissue macrophages develop from precursors that emerge later in life [103][104]. They constantly survey their milieu and assess ongoing synaptic activity, mediate synaptic pruning, clear debris, and provide trophic support for neurons [108]. In pathological situations such as chronic stress, the blood brain barrier (BBB) may become compromised, allowing peripheral hematopoietic cells to cross into neural tissue and become part of the parenchymal microglia/macrophage pool [109][110]. In response to CNS insults such as neuronal injury or infection, microglia become activated to produce pro-inflammatory factors (M1 phenotype) or anti-inflammatory factors (M2 phenotype) [111]. An exquisite balance of anti-inflammatory mediators to heal and repair tissues and pro-inflammatory mediators to clear cellular debris and aggregated misfolded proteins is essential for the maintenance of a healthy CNS [27][111]. With advancing age, microglia acquire an activated phenotype and release pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-6 [112][113][114]. In AD, microglia react to pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) [115][116][117] to assume a M1 phenotype, leading to an exacerbation of inflammation and an acceleration of disease progression [88] (Figure 1). Preliminary findings implicate a link between Aβ and neuroinflammation [118]. In AD brain slices, activated microglia surround both extracellular Aβ plaques and neurons containing neurofibrillary tangles (NFTs) [119][120]. It is thought that Aβ activate microglia, which then secrete IL1β, IL6, and TNFα, as well as (C-C motif) ligand (CCL 2/4/11), which lead to the recruitment of more microglia and astrocytes to the Aβ locus [121]. Microglia phagocytize Aβ through a range of cell surface receptors, including cluster of differentiation (CD)-14, toll-like receptor (TLR)-2, TLR4, α6β1 integrin, CD47, and scavenger receptors such as CD36 [122][123][124][125]. In AD, the accumulation of Aβ throughout the brain results partly from the failure of microglia to remove extracellular Aβ [126][127][128] and AD cortical specimens reveal that the microglia surrounding plaques have impaired Aβ uptake [127][129][130]. It has been shown in human and animal studies that inflammation influences APP processing overall [131][132]. Initially, microglial activation may serve to eliminate Aβ [126][133][134][135], but their chronic activation may amplify the amyloid cascade [133][136] and lead to neurotoxicity [137][138]. In rat intraventricular hemorrhage (IVH) model of AD, Aβ accumulation tracks with neuroinflammation and may contribute to the cognitive impairment [139].There is no consensus, however, regarding the relationship between in vivo microglial activation and Aβ plaque burden [140][141][142][143][144]. Aβ has recently been suggested to be an antimicrobial peptide that fibrilizes in order to activate the innate immune defense system and protect the host from a wide range of infectious agents [145].
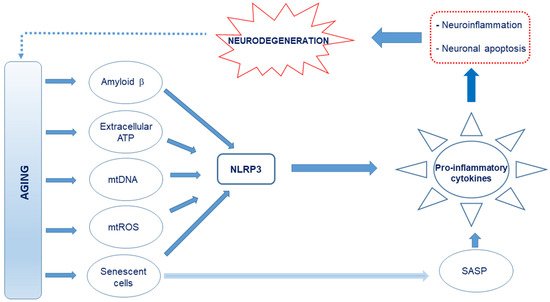
Figure 1. Schematic representation of the proposed causes of neuroinflammation in Alzheimer’s disease (AD). Age-related release of damage-associated molecular patterns (DAMPs) such as Aβ, extracellular ATP, and cell debris such as circulating mitochondrial DNA, which are capable of interacting with the Nod-like receptor 3 (NLRP3), creates an oxidative and neuroinflammatory environment through the excessive production and release of pro-inflammatory cytokines and reactive oxygen and nitrogen species (RONS). Further, mitochondrial reactive oxygen species (mtROS) and senescence-associated secretory phenotype (SASP) factors from the senescent cells, which also drive senescence in nearby cells, produce pro-inflammatory cytokines. This culminates in neuroinflammation and neuronal apoptosis.
2.2. Astrocytes
Astrocytes are the most abundant cell type in the CNS and a critical part of the tripartite synapse [146]. They are highly sensitive to their environment and rapidly respond to CNS needs and insults [147]. They also regulate the maturation of neurons and help maintain their function [147][148]. They are found in various states of activation and can be neuroprotective (reducing inflammation and stimulating repair) or neurotoxic (promoting inflammation that may result in neurodegeneration) [149][150]. They respond to inflammatory molecules such as cytokines and chemokines and are able to detect aggregated proteins such as Aβ [148][151][152]. They hypertrophy upon activation and upregulate glial fibrillary acidic protein (GFAP) expression [153][154]. Reactive astrocytes are a distinct trait of AD patient brains [155] and are also a feature of AD mouse model brains [156][157][158]. Intralaminar astrocytes are atrophied and severely disrupted in post-mortem AD brains [159]. In the 3xTg, which contain three mutations associated with a familial AD (APP Swedish, MAPT P301L, and PSEN1 M146V) mouse model, atrophic astrocytes appear in the entorhinal cortex (EC) as early as 30 days and are present until Aβ plaques begin to emerge at 12 months of age [160]. This phenomenon also occurs in other AD mouse models including the 5xTG-AD, PDAPP-J20, and Swiss 3 mice [161][162][163][164]. When astrocytes are created from familial and sporadic AD-induced pluripotent stem cells (iPSC), they have an atrophic phenotype in vitro [165]. Inhibiting astrogliosis in AD mouse brains results in Aβ accumulation with increased histopathology [166] and they are associated with cognition [167]. This may result in a breach of the blood brain barrier (BBB), leading to an infiltration of peripheral immune cells, aggravating neuroinflammation and inducing neurotoxicity by impairing glutamate homeostasis [168][169], and generating altered Ca2+ signaling [170].
2.3. Oligodendrocytes
The main function of oligodendrocytes is to provide support and insulation to the axons by forming myelin sheaths around nerve fibers. Their involvement in AD is not fully understood, although emerging evidence implicates their potential role in pathogenesis and progression of AD [171]. Tsai et al. recently reported that oligodendrocytes are severely impaired in AD [172] and, indeed, there is focal loss of oligodendrocytes and a reduction in myelin proteins near Aβ plaques [173][174][175]. Aβ not only impairs the survival and maturation of oligodendrocyte progenitor cells (OPCs), but also hampers the formation of the myelin sheath [176]. Neuroinflammation and oxidative stress may also contribute to oligodendrocyte dysfunction and death [175].
2.4. Myeloid Cells Other Than Microglia
The other monocytic cells found in the CNS include perivascular macrophages that line blood vessels of the brain, macrophages within the choroid plexus, and meningeal macrophages in the leptomeninges [177]. Dendritic cells, monocytes, and granulocytes are found in the meninges and are recruited to the brain during or after an insult or other pathology [178][179]. The CNS-resident macrophages express scavenger receptors (SR) and TLRs that facilitate phagocytosis and degradation of Aβ [180][181]. In SR knock out mouse models of AD, Aβ accumulates in the parenchyma and the animals have cognitive deficits [124][182]. In the CD11c-DNR mouse model of AD, which expresses a dominant-negative form of the TGF-β receptor under the control of the CD11c promoter, the brain levels of Aβ are reduced by up to 90%, the Aβ plaques and cerebral vasculature are surrounded by macrophages [183], and the behavior of the animals is significantly improved [183]. The migration of peripheral monocytes is dependent on C-C chemokine receptor type 2 (CCR2) [184]. Blocking transforming growth factor (TGF)-β signaling increases peripheral myeloid cell infiltration into the CNS and significantly reduces Aβ burden [183]. It is still not exactly clear how myeloid infiltration into the brain contributes to damage or clearance of pathological proteins.
3. Defective Autophagy and Neuroinflammation
Cells degrade protein aggregates and damaged organelles by autophagy and defective mitochondria by mitophagy [185][186][187][188]. With advancing age, autophagy gradually subsides and this decline is linked to defective mitochondria and results in inflammaging [189]. Damaged cellular and organelle components that accumulate as a result of inadequate autophagy are released as damage-associated molecular patterns (DAMPs) [190][191][192]. Dysfunctional mitochondria that are not eliminated by mitophagy release large amounts of mitochondrial DNA (mtDNA) into the cytosol and, together with ROS [193][194], metabolites such as ATP, fatty acids, Aβ, succinate, per-oxidized lipids, advanced glycation end-products, altered N-glycans, and HMGB1 are also recognized as DAMPs and trigger an innate immune inflammatory response [195][196] by directly activating TLR9. This initiates the transcription of pro-inflammatory cytokines such as IL-6, TNF-α, IL-1β, and MMP-8 [197] and activates the Nod-like receptor 3 (NLRP3) inflammasome, a key regulator of inflammation [198][199][200], to activate caspase-1 and facilitate IL-1β and IL-18 maturation as well as gasdermin D-mediated pyroptotic cell death [201][202][203]. These inflammatory responses can be blocked by Pro-IL-1β degradation in autophagosomes [204]. Further, the mitochondrial derived peptide (MDP) known as mitochondrial open reading frame of the 12S ribosomal RNA type-c (MOTS-c) [205] reduces inflammation by inhibiting cytokines such as TNF-α and IL-6, while simultaneously promoting an anti-inflammatory response. MOTS-c stimulates IL-10 as well as signal transducer and activators of transcription 3 (STAT3) and aryl hydrocarbon receptor (Ahr), which inhibit NFκB expression and proinflammatory cytokine production [206]. Another mitochondrial peptide, humanin, also has anti-inflammatory effects [207][208]. The chronic sterile low-grade inflammation elicited [4] may culminate in immunosenescence [209] and compromise neuronal function [32]. This may partly explain why dysregulated NLRP3 inflammasome activation is observed in AD [210][211]). Eliminating damaged and dysfunctional mitochondria by mitophagy may prevent the hyperinflammation triggered by NLRP3 inflammasome activation [212].
References
- Pattabiraman, G.; Palasiewicz, K.; Galvin, J.P.; Ucker, D.S. Aging-associated dysregulation of homeostatic immune response termination (and not initiation). Aging Cell 2017, 16, 585–593.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217.
- Spinelli, R.; Parrillo, L.; Longo, M.; Florese, P.; Desiderio, A.; Zatterale, F.; Miele, C.; Raciti, G.A.; Beguinot, F. Molecular basis of ageing in chronic metabolic diseases. J. Endocrinol. Investig. 2020, 43, 1373–1389.
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254.
- Li, T.; Huang, Y.; Cai, W.; Chen, X.; Men, X.; Lu, T.; Wu, A.; Lu, Z. Age-related cerebral small vessel disease and inflammaging. Cell Death Dis. 2020, 11, 1–12.
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95.
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960.
- Franceschi, C.; Santoro, A.; Capri, M. The complex relationship between Immunosenescence and Inflammaging: Special issue on the New Biomedical Perspectives. Semin. Immunopathol. 2020, 42, 517–520.
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247.
- Barbé-Tuana, F.; Funchal, G.; Schmitz, C.R.R.; Maurmann, R.M.; Bauer, M.E. The interplay between immunosenescence and age-related diseases. Semin. Immunopathol. 2020, 42, 545–557.
- Conte, M.; Martucci, M.; Chiariello, A.; Franceschi, C.; Salvioli, S. Mitochondria, immunosenescence and inflammaging: A role for mitokines? Semin. Immunopathol. 2020, 42, 607–617.
- Haas, R.H. Mitochondrial Dysfunction in Aging and Diseases of Aging. Biology 2019, 8, 48.
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175.
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. 2019, 9, 790.
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933.
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2021, 14.
- Lopez-Castejon, G. Control of the inflammasome by the ubiquitin system. FEBS J. 2020, 287, 11–26.
- Hegde, A.N.; Smith, S.G.; Duke, L.M.; Pourquoi, A.; Vaz, S. Perturbations of Ubiquitin-Proteasome-Mediated Proteolysis in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 324.
- Ioannidou, A.; Goulielmaki, E.; Garinis, G.A. DNA Damage: From Chronic Inflammation to Age-Related Deterioration. Front. Genet. 2016, 7, 187.
- da Silva, P.F.L.; Schumacher, B. DNA damage responses in ageing. Open Biol. 2019, 9, 190168.
- Chen, G.; Yung, R. Meta-inflammaging at the crossroad of geroscience. Aging Med. 2019, 2, 157–161.
- Herradon, G.; Ramos-Alvarez, M.P.; Gramage, E. Connecting Metainflammation and Neuroinflammation Through the PTN-MK-RPTPβ/ζ Axis: Relevance in Therapeutic Development. Front. Pharmacol. 2019, 10, 10.
- Vitale, G.; Salvioli, S.; Franceschi, C. Oxidative stress and the ageing endocrine system. Nat. Rev. Endocrinol. 2013, 9, 228–240.
- Shintouo, C.M.; Mets, T.; Beckwee, D.; Bautmans, I.; Ghogomu, S.M.; Souopgui, J.; Leemans, L.; Meriki, H.D.; Njemini, R. Is inflammageing influenced by the microbiota in the aged gut? A systematic review. Exp. Gerontol. 2020, 141, 111079.
- Santoro, A.; Zhao, J.; Wu, L.; Carru, C.; Biagi, E.; Franceschi, C. Microbiomes other than the gut: Inflammaging and age-related diseases. Semin. Immunopathol. 2020, 42, 589–605.
- Tang, Y.; Fung, E.; Xu, A.; Lan, H.-Y. C-reactive protein and ageing. Clin. Exp. Pharmacol. Physiol. 2017, 44, 9–14.
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586.
- Marcos-Pérez, D.; Sánchez-Flores, M.; Proietti, S.; Bonassi, S.; Costa, S.; Teixeira, J.P.; Fernández-Tajes, J.; Pásaro, E.; Laffon, B.; Valdiglesias, V. Association of inflammatory mediators with frailty status in older adults: Results from a systematic review and meta-analysis. GeroScience 2020, 42, 1451–1473.
- Tchalla, A.E.; Wellenius, G.A.; Travison, T.G.; Gagnon, M.; Iloputaife, I.; Dantoine, T.; Sorond, F.A.; Lipsitz, L.A. Circulating Vascular Cell Adhesion Molecule-1 Is Associated With Cerebral Blood Flow Dysregulation, Mobility Impairment, and Falls in Older Adults. Hypertension 2015, 66, 340–346.
- Prochaska, J.H.; Frank, B.; Nagler, M.; Lamparter, H.; Weißer, G.; Schulz, A.; Eggebrecht, L.; Göbel, S.; Arnold, N.; Panova-Noeva, M.; et al. Age-related diagnostic value of D-dimer testing and the role of inflammation in patients with suspected deep vein thrombosis. Sci. Rep. 2017, 7, 1–10.
- Lee, S.-H.; Lee, J.-H.; Lee, H.-Y.; Min, A.K.-J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34.
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 4–9.
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713.
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396.
- Gross, A.L.; Walker, K.A.; Moghekar, A.R.; Pettigrew, C.; Soldan, A.; Albert, M.S.; Walston, J.D. Plasma Markers of Inflammation Linked to Clinical Progression and Decline During Preclinical AD. Front. Aging Neurosci. 2019, 11, 229.
- Kuo, C.-Y.; Stachiv, I.; Nikolai, T. Association of Late Life Depression, (Non-) Modifiable Risk and Protective Factors with Dementia and Alzheimer’s Disease: Literature Review on Current Evidences, Preventive Interventions and Possible Future Trends in Prevention and Treatment of Dementia. Int. J. Environ. Res. Public Health 2020, 17, 7475.
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581.
- Cummings, J. New approaches to symptomatic treatments for Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 1–13.
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33.
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32.
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056.
- Roda, A.R.; Montoliu-Gaya, L.; Serra-Mir, G.; Villegas, S. Both Amyloid-β Peptide and Tau Protein Are Affected by an Anti-Amyloid-β Antibody Fragment in Elderly 3xTg-AD Mice. Int. J. Mol. Sci. 2020, 21, 6630.
- Reitz, C.; Rogaeva, E.; Beecham, G.W. Late-onset vs nonmendelian early-onset Alzheimer disease: A distinction without a difference? Neurol. Genet. 2020, 6, e512.
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651.
- Cruchaga, C.; Del-Aguila, J.L.; Saef, B.; Black, K.; Fernandez, M.V.; Budde, J.; Ibanez, L.; Deming, Y.; Kapoor, M.; Tosto, G.; et al. Polygenic risk score of sporadic late-onset Alzheimer’s disease reveals a shared architecture with the familial and early-onset forms. Alzheimer’s Dement. 2018, 14, 205–214.
- Kelleher, R.J., 3rd; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631.
- A An, S.S.; Cai, Y.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 2015, 10, 1163–1172.
- Tambini, M.D.; A Norris, K.; D’Adamio, L. Opposite changes in APP processing and human Aβ levels in rats carrying either a protective or a pathogenic APP mutation. eLife 2020, 9, 9.
- Vidal, C.; Zhang, L. An Analysis of the Neurological and Molecular Alterations Underlying the Pathogenesis of Alzheimer’s Disease. Cells 2021, 10, 546.
- Tang, M.; Ryman, D.C.; McDade, E.; Jasielec, M.S.; Buckles, V.D.; Cairns, N.J.; Faga, A.M.; Goate, A.; Marcus, D.S.; Xiong, C.; et al. Neurological manifestations of autosomal dominant familial Alzheimer’s disease: A comparison of the published literature with the dominantly inherited Alzheimer network observational study (DIAN-OBS). Lancet Neurol. 2016, 15, 1317–1325.
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106.
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923.
- Naj, A.C.; Schellenberg, G.D. Alzheimer’s Disease Genetics Consortium (ADGC). Genomic variants, genes, and pathways of Alzheimer’s disease: An overview. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 5–26.
- Prokopenko, D.; Morgan, S.L.; Mullin, K.; Hofmann, O.; Chapman, B.; Kirchner, R.; Amberkar, S.; Wohlers, I.; Lange, C.; Hide, W.; et al. Whole-genome sequencing reveals new Alzheimer’s disease–associated rare variants in loci related to synaptic function and neuronal development. Alzheimer’s Dement. 2021.
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51.
- Grozeva, D.; Saad, S.; Menzies, G.E.; Sims, R. Benefits and Challenges of Rare Genetic Variation in Alzheimer’s Disease. Curr. Genet. Med. Rep. 2019, 7, 53–62.
- Lord, J.; Lu, A.J.; Cruchaga, C. Identification of rare variants in Alzheimer’s disease. Front. Genet. 2014, 5, 369.
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458.
- Sims, R.; van der Lee, S.J.; Naj, A.C.; Bellenguez, C.; Badarinarayan, N.; Jakobsdottir, J.; Kunkle, B.W.; Boland, A.; Raybould, R.; Bis, J.C.; et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017, 49, 1373–1384.
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12.
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–16.
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772.
- Klein, H.-U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019, 22, 37–46.
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 2018, 98, 1141–1154.
- Gambhir, I.; Misra, A.; Chakrabarti, S. New genetic players in late-onset Alzheimer’s disease: Findings of genome-wide association studies. Indian J. Med Res. 2018, 148, 135–144.
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608.
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.G.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127.
- Li, J.-T.; Zhang, Y. TREM2 regulates innate immunity in Alzheimer’s disease. J. Neuroinflamm. 2018, 15.
- Zheng, H.; Cheng, B.; Li, Y.; Li, X.; Chen, X.; Zhang, Y.-W. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 2018, 10, 395.
- Zhong, L.; Chen, X.-F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607.
- Hickman, S.E.; El Khoury, J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 495–498.
- Gong, C.-X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, S107–S117.
- De Roeck, A.; Van Broeckhoven, C.; Sleegers, K. The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019, 138, 201–220.
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimer's Dement. 2010, 6, 420–424.
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372.
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673.
- Shen, Z.; Bao, X.; Wang, R. Clinical PET Imaging of Microglial Activation: Implications for Microglial Therapeutics in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 314.
- Yao, K.; Zu, H.B. Microglial polarization: Novel therapeutic mechanism against Alzheimer’s disease. Inflammopharmacology 2020, 28, 95–110.
- Tondo, G.; Iaccarino, L.; Caminiti, S.P.; Presotto, L.; Santangelo, R.; Iannaccone, S.; Magnani, G.; Perani, D. The combined effects of microglia activation and brain glucose hypometabolism in early-onset Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 1–10.
- Wang, W.-Y.; Tan, M.-S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136.
- Stamouli, E.C.; Politis, A.M. Pro-inflammatory cytokines in Alzheimer’s disease. Psychiatriki 2016, 27, 264–275.
- Wang, M.-M.; Miao, D.; Cao, X.-P.; Tan, L.; Tan, L. Innate immune activation in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 177.
- Fernández-Arjona, M.D.M.; Grondona, J.M.; Fernández-Llebrez, P.; López-Ávalos, M.D. Microglial Morphometric Parameters Correlate With the Expression Level of IL-1β, and Allow Identifying Different Activated Morphotypes. Front. Cell Neurosci. 2019, 13, 472.
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421.
- Azizi, G.; Navabi, S.S.; Al-Shukaili, A.; Seyedzadeh, M.H.; Yazdani, R.; Mirshafiey, A. The Role of Inflammatory Mediators in the Pathogenesis of Alzheimer’s Disease. Sultan Qaboos Univ. Med. J. 2015, 15, e305–e316.
- Chen, X.Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 659.
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405.
- Rios, M.A.E.; Etoral-Rios, D.; Efranco-Bocanegra, D.; Evilleda-Hernández, J.; Ecampos-Peña, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59.
- Businaro, R.; Corsi, M.; Asprino, R.; Di Lorenzo, C.; Laskin, D.; Corbo, R.; Ricci, S.; Pinto, A. Modulation of Inflammation as a Way of Delaying Alzheimer’s Disease Progression: The Diet’s Role. Curr. Alzheimer Res. 2018, 15, 363–380.
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell Neurosci. 2018, 12, 72.
- Duran-Aniotz, C.; Hetz, C. Glucose Metabolism: A Sweet Relief of Alzheimer’s Disease. Curr. Biol. 2016, 26, R806–R809.
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer's Dis. 2017, 57, 1105–1121.
- Thal, D.R.; Capetillo-Zarate, E.; Larionov, S.; Staufenbiel, M.; Zurbruegg, S.; Beckmann, N. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol. Aging 2009, 30, 1936–1948.
- Potter, H.; Granic, A.; Caneus, J. Role of Trisomy 21 Mosaicism in Sporadic and Familial Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 13, 7–17.
- Clare, R.; King, V.G.; Wirenfeldt, M.; Vinters, H.V. Synapse loss in dementias. J. Neurosci. Res. 2010, 88, 2083–2090.
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 1–22.
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019, 216, 60–70.
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746.
- Buttgereit, A.; Lelios, I.; Yu, X.; Vrohlings, M.; Krakoski, N.R.; Gautier, E.L.; Nishinakamura, R.; Becher, B.; Greter, M. Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 2016, 17, 1397–1406.
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2014, 518, 547–551.
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340.
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 2015, 43, 382–393.
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678.
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543.
- Tay, T.L.; Hagemeyer, N.; Prinz, M. The force awakens: Insights into the origin and formation of microglia. Curr. Opin. Neurobiol. 2016, 39, 30–37.
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397.
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698.
- Mildner, A.; Schmidt, H.; Nitsche, M.; Merkler, D.; Hanisch, U.-K.; Mack, M.; Heikenwalder, M.; Brück, W.; Priller, J.; Prinz, M.; et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007, 10, 1544–1553.
- Wohleb, E.S.; Powell, N.D.; Godbout, J.P.; Sheridan, J.F. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J. Neurosci. 2013, 33, 13820–13833.
- Sochoka, M.; Diniz, B.S.; Leszek, J. Inflammatory responses in the CNS: Fried or foe? Mol. Neurobiol. 2017, 54, 8071–8089.
- Tejera, D.; Heneka, M.T. Microglia in Alzheimer’s disease: The good, the bad and the ugly. Curr. Alzheimer Res. 2016, 13, 370–380.
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harbor Perspect. Med. 2012, 2, a006346.
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W.G.M. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602.
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748.
- Koenigsknecht-Talboo, J.; Landreth, G.E. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 2005, 25, 8240–8249.
- Sondag, C.M.; Dhawan, G.; Combs, C.K. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J. Neuroinflamm. 2009, 6.
- Hommet, C.; Mondon, K.; Camus, V.; Ribeiro, M.J.; Beaufils, E.; Arlicot, N.; Corcia, P.; Paccalin, M.; Minier, F.; Gosselin, T.; et al. Neuroinflammation and β Amyloid Deposition in Alzheimer’s Disease: In vivo Quantification with Molecular Imaging. Dement. Geriatr. Cogn. Disord. 2014, 37, 1–18.
- Nagele, R.G.; Wegiel, J.; Venkataraman, V.; Imaki, H.; Wang, K.C.; Wegiel, J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 663–674.
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2020, 179, 1373–1384.
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474.
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674.
- Ries, M.; Sastre, M. Mechanisms of Aβ clearance and degradation by glial cells. Front. Aging Neurosci. 2016, 8, 160.
- Tahara, K.; Kim, H.-D.; Jin, J.-J.; Maxwell, J.A.; Li, L.; Fukuchi, K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain J. Neurol. 2006, 129, 3006–3019.
- Wilkinson, K.; El Khoury, J. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer’s disease. Int. J. Alzheimer's Dis. 2012, 2012, 489456.
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360.
- Thériault, P.; ElAli, A.; Rivest, S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimer's Res. Ther. 2015, 7.
- Weiner, H.L.; Frenkel, D. Immunology and immunotherapy of Alzheimer’s disease. Nat. Rev. Immunol. 2006, 6, 404–416.
- Frackowiak, J.; Wisniewski, H.M.; Wegiel, J.; Merz, G.S.; Iqbal, K.; Wang, K.C. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992, 84, 225–233.
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 2013, 8, e60921.
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003, 971, 197–209.
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453.
- Meda, L.; Baron, P.; Scarlato, G. Glial activation in Alzheimer’s disease: The role of Abeta and its associated proteins. Neurobiol. Aging 2001, 22, 885–893.
- Morgan, D.; Gordon, M.N.; Tan, J.; Wilcock, D.; Rojiani, A.M. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: Implications for Alzheimer therapeutics. J. Neuropathol. Exp. Neurol. 2005, 64, 743–753.
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305.
- Tan, Z.S.; Seshadri, S. Inflammation in the Alzheimer’s disease cascade: Culprit or innocent bystander? Alzheimer's Res. Ther. 2010, 2, 6.
- Joshi, P.G.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593.
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472.
- Jiang, C.; Zou, X.; Zhu, R.; Shi, Y.; Wu, Z.; Zhao, F.; Chen, L. The correlation between accumulation of amyloid beta with enhanced neuroinflammation and cognitive impairment after intraventricular hemorrhage. J. Neurosurg. 2018, 131, 54–63.
- Edison, P.; Archer, H.A.; Gerhard, A.; Hinz, R.; Pavese, N.; Turkheimer, F.E.; Hammers, A.; Tai, Y.F.; Fox, N.; Kennedy, A.; et al. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol. Dis. 2008, 32, 412–419.
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.; Kennedy, J.; Bullock, R.; Walker, Z.; Fox, N.; Rossor, M.; Brooks, D.J. Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology 2009, 72, 56–62.
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; De Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using18F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264.
- Fan, Z.; Okello, A.A.; Brooks, D.J.; Edison, P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain 2015, 138, 3685–3698.
- Yokokura, M.; Mori, N.; Yagi, S.; Yoshikawa, E.; Kikuchi, M.; Yoshihara, Y.; Wakuda, T.; Sugihara, G.; Takebayashi, K.; Suda, S.; et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2010, 38, 343–351.
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimer's Dis. 2018, 62, 1495–1506.
- Chung, W.-S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370.
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389.
- Preman, P.; Alfonso-Triguero, M.; Alberdi, E.; Verkhratsky, A.; Arranz, A. Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways. Cells 2021, 10, 540.
- Sheeler, C.; Rosa, J.-G.; Ferro, A.; McAdams, B.; Borgenheimer, E.; Cvetanovic, M. Glia in Neurodegeneration: The Housekeeper, the Defender and the Perpetrator. Int. J. Mol. Sci. 2020, 21, 9188.
- Gamage, R.; Wagnon, I.; Rossetti, I.; Childs, R.; Niedermayer, G.; Chesworth, R.; Gyengesi, E. Cholinergic Modulation of Glial Function During Aging and Chronic Neuroinflammation. Front. Cell. Neurosci. 2020, 14, 577912.
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622.
- Giovannoni, F.; Quintana, F.J. The role of astrocytes in CNS inflammation. Trends Immunol. 2020, 41, 805–819.
- Hol, E.M.; Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 2015, 32, 121–130.
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374.
- Beach, T.; McGeer, E. Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer’s disease visual cortex. Brain Res. 1988, 463, 357–361.
- Rodríguez, J.J.; Olabarria, M.; Chvatal, A.; Verkhratsky, A.; Rodr, J.J. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 2008, 16, 378–385.
- Verkhratsky, A.; Zorec, R.; Parpura, V. Stratification of astrocytes in healthy and diseased brain. Brain Pathol. 2017, 27, 629–644.
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427.
- Colombo, J.; Quinn, B.; Puissant, V. Disruption of astroglial interlaminar processes in Alzheimer’s disease. Brain Res. Bull. 2002, 58, 235–242.
- Yeh, C.-Y.; Vadhwana, B.; Verkhratsky, A.; Rodriguez, J.J. Early Astrocytic Atrophy in the Entorhinal Cortex of a Triple Transgenic Animal Model of Alzheimer’s Disease. ASN Neuro 2011, 3, e00071.
- Beauquis, J.; Vinuesa, A.; Pomilio, C.; Pavía, P.; Galván, V.; Saravia, F. Neuronal and Glial Alterations, Increased Anxiety, and Cognitive Impairment before Hippocampal Amyloid Deposition in PDAPP Mice, Model of Alzheimer’s Disease. Hippocampus 2014, 24, 257–269.
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Araujo, A.P.B.; Melo, H.M.; Da Silva, G.S.S.; Alves-Leon, S.V.; De Souza, J.M.; Ferreira, S.T.; et al. Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Aβ Oligomers in Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 6797–6809.
- Iram, T.; Trudler, D.; Kain, D.; Kanner, S.; Galron, R.; Vassar, R.; Barzilai, A.; Blinder, P.; Fishelson, Z.; Frenkel, D. Astrocytes from old Alzheimer’s disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol. Dis. 2016, 96, 84–94.
- Polis, B.; Srikanth, K.D.; Elliott, E.; Gil-Henn, H.; Samson, A.O. L-Norvaline Reverses Cognitive Decline and Synaptic Loss in a Murine Model of Alzheimer’s Disease. Neurotherapeutics 2018, 15, 1036–1054.
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant iPSC-Derived Human Astrocytes in Alzheimer’s Disease. Cell Death Dis. 2017, 8, e2696.
- Kraft, A.W.; Hu, X.; Yoon, H.; Yan, P.; Xiao, Q.; Wang, Y.; Gil, S.C.; Brown, J.; Wilhelmsson, U.; Restivo, J.L.; et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013, 27, 187–198.
- Kobayashi, E.; Nakano, M.; Kubota, K.; Himuro, N.; Mizoguchi, S.; Chikenji, T.; Otani, M.; Mizue, Y.; Nagaishi, K.; Fujimiya, M. Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep. 2018, 8, 1–12.
- Masliah, E.; Alford, M.; DeTeresa, R.; Mallory, M.; Hansen, L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766.
- Scimemi, A.; Meabon, J.S.; Woltjer, R.L.; Sullivan, J.M.; Diamond, J.S.; Cook, D.G. Amyloid-β1-42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J. Neurosci. 2013, 33, 5312–5318.
- Vincent, A.J.; Gasperini, R.; Foa, L.; Small, D.H. Astrocytes in Alzheimer’s Disease: Emerging Roles in Calcium Dysregulation and Synaptic Plasticity. J. Alzheimer’s Dis. 2010, 22, 699–714.
- Ettle, B.; Schlachetzki, J.C.M.; Winkler, J. Oligodendroglia and myelin in neurodegenerative diseases: More than just bystanders? Mol. Neurobiol. 2016, 53, 3046–3062.
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337.
- Bartzokis, G. Age-related myelin breakdown: A developmental model of cognitive decline and Alzheimer’s disease. Neurobiol. Aging 2004, 25, 5–18.
- Bartzokis, G.; Lu, P.H.; Mintz, J. Quantifying age-related myelin breakdown with MRI: Novel therapeutic targets for preventing cognitive decline and Alzheimer’s disease. J. Alzheimer's Dis. 2004, 6, S53–S59.
- Mitew, S.; Kirkcaldie, M.T.K.; Halliday, G.M.; Shepherd, C.E.; Vickers, J.C.; Dickson, T.C. Focal demyelination in Alzheimer’s disease and transgenic mouse models. Acta Neuropathol. 2010, 119, 567–577.
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early Oligodendrocyte/Myelin Pathology in Alzheimer’s Disease Mice Constitutes a Novel Therapeutic Target. Am. J. Pathol. 2010, 177, 1422–1435.
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805.
- Chinnery, H.R.; Ruitenberg, M.J.; McMenamin, P.G. Novel characterization of monocyte-derived cell populations in the meninges and choroid plexus and their rates of replenishment in bone marrow chimeric mice. J. Neuropathol. Exp. Neurol. 2010, 69, 896–909.
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312.
- Mohamed, A.; Posse de Chaves, E. Aβ internalization by neurons and glia. Int. J. Alzheimer's Dis. 2011, 2011, 127984.
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.-P.; Rivest, S. Bone Marrow-Derived Microglia Play a Critical Role in Restricting Senile Plaque Formation in Alzheimer’s Disease. Neuron 2006, 49, 489–502.
- Thanopoulou, K.; Fragkouli, A.; Stylianopoulou, F.; Georgopoulos, S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 20816–20821.
- Town, T.; Laouar, Y.; Pittenger, C.; Mori, T.; Szekely, C.A.; Tan, J.; Duman, R.S.; Flavell, R.A. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 2008, 14, 681–687.
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438.
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22.
- Li, W.; He, P.; Huang, Y.; Li, Y.-F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256.
- Wang, R.; Wang, G. Autophagy in Mitochondrial Quality Control. Adv. Exp. Med. Biol. 2019, 1206, 421–434.
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108.
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the Autophagy-Inflammation-Cell Death Axis in Organismal Aging. Science 2011, 333, 1109–1112.
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. Mol. Life Sci. 2014, 157, 65–75.
- Desai, S.; Juncker, M.; Kim, C. Regulation of mitophagy by the ubiquitin pathway in neurodegenerative diseases. Exp. Biol. Med. 2018, 243, 554–562.
- Madruga, E.; Maestro, I.; Martínez, A. Mitophagy Modulation, a New Player in the Race against ALS. Int. J. Mol. Sci. 2021, 22, 740.
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230.
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414.
- Dall’Olio, F.; Vanhooren, V.; Chen, C.C.; Slagboom, P.E.; Wuhrer, M.; Franceschi, C. N-glycomic biomarkers of biological aging and longevity: A link with inflammaging. Ageing Res. Rev. 2013, 12, 685–698.
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39.
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 2016, 7, 11–16.
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022.
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315.
- Mamik, M.K.; Power, C. Inflammasomes in neurological diseases: Emerging pathogenic and therapeutic concepts. Brain 2017, 140, 2273–2285.
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489.
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Asp. Med. 2020, 76, 100924.
- Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014, 35, 253–261.
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1 secretion by targeting pro-IL-1 for degradation. J. Biol. Chem. 2011, 286, 9587–9597.
- Lee, C.; Kim, K.H.; Cohen, P. MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free. Radic. Biol. Med. 2016, 100, 182–187.
- Zhai, D.; Ye, Z.; Jiang, Y.; Xu, C.; Ruan, B.; Yang, Y.; Lei, X.; Xiang, A.; Lu, H.; Zhu, Z.; et al. MOTS-c peptide increases survival and decreases bacterial load in mice infected with MRSA. Mol. Immunol. 2017, 92, 151–160.
- Oh, Y.K.; Bachar, A.R.; Zacharias, D.G.; Kim, S.G.; Wan, J.; Cobb, L.J.; Lerman, L.O.; Cohen, P.; Lerman, A. Humanin preserves endothelial function and prevents atherosclerotic plaque progression in hypercholesterolemic ApoE deficient mice. Atherosclerosis 2011, 219, 65–73.
- Sreekumar, P.G.; Ishikawa, K.; Spee, C.; Mehta, H.H.; Wan, J.; Yen, K.; Cohen, P.; Kannan, R.; Hinton, D.R. The Mitochondrial-Derived Peptide Humanin Protects RPE Cells From Oxidative Stress, Senescence, and Mitochondrial Dysfunction. Investig. Opthalmology Vis. Sci. 2016, 57, 1238–1253.
- Muzumdar, R.H.; Huffman, D.M.; Atzmon, G.; Buettner, C.; Cobb, L.J.; Fishman, S.; Budagov, T.; Cui, L.; Einstein, F.H.; Poduval, A.; et al. Humanin: A Novel Central Regulator of Peripheral Insulin Action. PLoS ONE 2009, 4, e6334.
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865.
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236.
- Kim, M.-J.; Yoon, J.-H.; Ryu, J.-H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016, 49, 529–535.

