 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kaschin Jamal Jameel | + 9313 word(s) | 9313 | 2021-07-09 10:39:32 | | | |
| 2 | Amina Yu | Meta information modification | 9313 | 2021-07-13 03:07:15 | | | | |
| 3 | Kaschin Jamal Jameel | -6346 word(s) | 2967 | 2021-07-13 03:13:32 | | | | |
| 4 | Amina Yu | -1383 word(s) | 1584 | 2021-07-14 03:56:58 | | | | |
| 5 | Amina Yu | Meta information modification | 1584 | 2021-07-14 09:45:37 | | |
Video Upload Options
In smoking-induced chronic obstructive pulmonary disease (COPD), various comorbidities are linked to systemic inflammation and infection-induced exacerbations. The underlying mechanisms are unclear but might provide therapeutic targets. T-cell activity is central in systemic inflammation and for infection-defense mechanisms and might be influenced by comorbidities. Hypothesis: Circulating biomarkers of comorbidities modulate the activity of T-cells of the T-helper type 1 (Th1) and/or T-cytotoxic type 1 (Tc1). T-cells in peripheral blood mononuclear cells (PBMCs) from non-smokers (NS), current smokers without COPD (S), and COPD subjects (total n = 34) were ex vivo activated towards Th1/Tc1 and were then stimulated with biomarkers for metabolic and/or cardiovascular comorbidities (Brain Natriuretic Peptide, BNP; chemokine (C-C motif) ligand 18, CCL18; C-X3-C motif chemokine ligand 1, CX3CL1; interleukin-18, IL-18) or for asthma- and/or cancer-related comorbidities (CCL22; epidermal growth factor, EGF; IL-17; periostin) each at 10 or 50 ng/mL. The Th1/Tc1 activation markers interferon-γ (IFNγ), tumor necrosis factor-α (TNFα), and granulocyte-macrophage colony-stimulating factor (GM-CSF) were analyzed in culture supernatants by Enzyme-Linked Immunosorbent Assay (ELISA). Ex-vivo activation induced IFNγ and TNFα without differences between the groups but GM-CSF more in S vs. NS. At 10 ng/mL, the different biomarkers increased or reduced the T-cell activation markers without a clear trend for one direction in the different categories of comorbidities or for the different T-cell activation markers. At 50 ng/mL, there was a clear shift towards suppressive effects, particularly for the asthma— and cancer-related biomarkers and in cells of S and COPD. Comorbidities might suppress T-cell immunity in COPD. This could explain the association of comorbidities with frequent exacerbations.
1. Introduction
Chronic obstructive pulmonary disease (COPD) is mainly induced by tobacco smoking. It is a systemic inflammatory disease associated with various comorbidities that have a negative impact on prognosis and progression. The development and progression of comorbidities might be triggered by systemic inflammation[1]. Comorbidities are associated with the frequency of exacerbations, another major trigger of progression[2]. COPD subjects have an increased susceptibility to bacterial and viral infections, both of which are major causes of exacerbations [3].
2. T-cell Activity and Comorbidities
Mechanistic links between T-cell activity and comorbidities are largely unknown but might provide insights into the association of comorbidities with exacerbations and suggest corresponding drug targets. Many comorbidities are characterized by circulating biomarkers that are often cytokines or chemokines whose receptors are expressed on T-cells or on accessory immune cells. In response to airway infections with most COPD-characteristic pathogens, inactive naive and memory T-cells become activated towards Th1 or Tc1 effector T-cells. During this activation process, the T-cells become recruited from the circulation to the local sites of infection. This implicates that comorbidities might influence T-cell activation via their circulating biomarkers, a possible mechanistic link between systemic inflammation, comorbidities and exacerbations. Therefore, this study aimed to investigate the impact of systemic biomarkers for frequent COPD comorbidities on T-cell activity
3. Frequent Comorbidities in COPD
Frequent comorbidities in COPD are metabolic and heart diseases, particularly type 2 diabetes, arteriosclerosis, coronary artery disease, and heart failure, all of which are linked to each other [4][5]. There is evidence from retrospective studies that comorbidities and particularly diabetes and heart failure are associated with frequent exacerbations in COPD [2]. The adipokine C-X3-C motif chemokine ligand 1 (CX3CL1, Fractalkine) and interleukin-18 (IL-18) both are associated with type 2 diabetes and are increased in the plasma of respective patients [6][7]. CX3CL1 and IL-18 both are further associated with coronary artery disease (CAD) and are increased in the plasma of CAD patients [8][9]. Chemokine (C-C motif) ligand 18 (CCL18) is also increased in the plasma of CAD patients and is associated with CAD severity [10]. CX3CL1 and CCL18, both are particularly increased in patients with refractory unstable angina, a symptom of CAD [9][11]. CX3CL1 is further associated with Carotid artery stenosis, a common coincidence with CAD and a further common consequence of arteriosclerosis, and is increased in the plasma of respective patients [12]. Brain Natriuretic Peptide (BNP) is a strong biomarker for heart failure, for which CAD is a major risk factor [13]. Its plasma levels are increased in patients with acute heart failure [14]. Serum CX3CL1, IL-18, and CCL18 are increased in stable COPD and are associated with disease progression and severity [15][16][17]. An association between BNP and stable COPD has, to our knowledge, not yet been observed. Further common comorbidities in COPD are asthma, lung cancer, and non-pulmonary cancer types like bladder, breast, colorectal, ovarian, and prostate cancer, for example [18][19]. Asthma and lung cancer might also be associated with frequent exacerbations in COPD [2]. An increased serum periostin level is a strong marker for type 2 asthma [20] and might also have prognostic significance for non-small cell lung cancer (NSCLC) and various non-pulmonary cancer types including those mentioned [21][22][23]. Serum IL-17 is increased in obesity-associated asthma and might be indicative of severe phenotypes [24][25]. Serum CCL22 is increased in breast cancer and is indicative of progression and severity [26]. Serum epidermal growth factor (EGF) is suitable to distinguish NSCLC from healthy benign lung pathologies [27]. Increased serum EGF concentrations are also discussed as putative biomarkers for various non-pulmonary cancer types including gastrointestinal cancers [28]. Serum periostin and EGF but not IL-17 and CCL22 are increased in stable COPD [29][30][31][32].4. Results
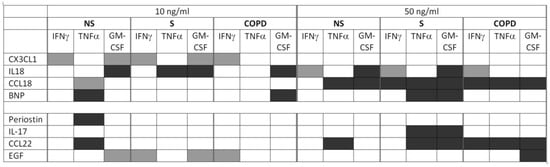
5. Discussion
To address the question of mechanistic links between co-morbidities, systemic inflammation and infection-induced exacerbations, we tested for effects of the respective circulating biomarkers on T-cell activity and T-cell activation towards Th1/Tc1 in the PBMC culture model. The model considers the presence of accessory cells that influence the T-cell activation process and the possible influence of co-morbidity biomarkers at recruitment from the circulation to the draining lymph nodes in response to acute infections in vivo. Independent of disease and smoking status of the subjects, the effects of a single biomarker concentration may vary in strength and also in direction between IFNγ, TNFα, and GM-CSF (Figure 1). At 10 ng/mL, the different biomarkers increased or reduced the T-cell activation markers without a clear trend for one direction in the different categories of comorbidities or for the different T-cell activation markers. However, increasing the biomarker concentrations clearly resulted in an increase of the suppressive effects, particularly for the biomarkers associated with asthma and cancer (Figure 1). Indeed, at 50 ng/mL, an up-regulation was only observed for IFNγ in response to the diabetes and CAD marker IL-18, but a reduction for at least one of the three T-cell activity markers in response to all other biomarkers except CX3CL1 and periostin. We interpret a reduced T-cell activity caused by a comorbidity-biomarker in our experimental model as a putative mechanistic reason for an increased possibility to get or prolong an exacerbation in COPD with the respective comorbidity. This is because a reduced T-cell-dependent infection defense might result in a delayed clearance of the pathogen. In this context, it is important to observe the respective biomarker effects in the COPD group but it is irrelevant if there are differences to healthy subjects or active smokers without respiratory symptoms. With two exceptions, we did not detect statistically significant differences when comparing the three subject groups, NS, S, and COPD, for the biomarker effects on T-cell activity. Nevertheless, we think that there is some evidence for an influence of active smoking and systemic COPD pathology on the biomarker effects. It is noteworthy that at 50 ng/mL statistically significant suppressive effects were more often observed in S and COPD than in NS, particularly again for those biomarkers associated with asthma and cancer (Figure 1). In NS, only TNFα was reduced by CCL22. In S, TNFα and GM-CSF both were reduced by CCL22 and by IL-17. In COPD, we did not find effects of IL-17, but all three T-cell activity markers were reduced by CCL22, and GM-CSF was also reduced by EGF.
This provides first evidence that systemic consequences of active smoking as well as of the COPD systemic pathology might influence the effects of asthma and cancer related co-morbidity biomarkers on T-cell activity. To a lesser extent this also applies to the biomarkers of cardiovascular co-morbidities, CCL18 and BNP. In summary, we carefully conclude that smoking and the COPD pathology might enhance the suppressive effects of the biomarkers for co-morbidities on T-cell immunity. However, because of the low numbers of significant differences between the groups, this conclusion requires further investigation. The observation that some of the biomarkers also modulate the T-cell activity in cells of healthy never smokers provides first indication that the diseases that are associated with the biomarkers might also influence T-cell immunity in the absence of COPD. However, compared to the COPD group, the overall trend was less pronounced towards suppressive effects. This study adds another part to the understanding of the complex systemic molecular pathology that underlies the increased susceptibility to respiratory infections in COPD. In contrast to the local innate immune cells that show an overactivation in response to respiratory pathogens in COPD [33][34], the activation process of circulating innate and adaptive immune cells appears to be rather suppressed. We have previously shown that systemic defects in Toll-like receptor signaling prevent the full activation of T-cells and monocytes in response to respiratory bacteria [35][36][37]. Here, we add the information that the suppression of the Th1-immunity might be amplified by co-morbidities in COPD.
References
- Pelgrim, C.E.; Peterson, J.D.; Gosker, H.R.; Schols, A.M.; van Helvoort, A.; Garssen, J.; Folkerts, G.; Kraneveld, A.D. Psychological co-morbidities in COPD: Targeting systemic inflammation, a benefit for both? Eur. J. Pharmacol. 2019, 842, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Westerik, J.A.M.; Metting, E.I.; Van Boven, J.F.M.; Tiersma, W.; Kocks, J.W.H.; Schermer, T.R. Associations between chronic comorbidity and exacerbation risk in primary care patients with COPD. Respir. Res. 2017, 18, 31. [Google Scholar] [CrossRef] [PubMed]
- Rangelov, K.; Sethi, S. Role of Infections. Clin. Chest Med. 2014, 35, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Gayle, A.; Dickinson, S.; Poole, C.; Pang, M.; Fauconnot, O.; Quint, J.K. Incidence of type II diabetes in chronic obstructive pulmonary disease: A nested case–control study. NPJ Prim. Care Respir. Med. 2019, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Trinkmann, F.; Saur, J.; Borggrefe, M.; Akin, I. Cardiovascular Comorbidities in Chronic Obstructive Pulmonary Disease (COPD)—Current Considerations for Clinical Practice. J. Clin. Med. 2019, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Hinkle, C.C.; Ferguson, J.; Mehta, N.N.; Li, M.; Qu, L.; Lu, Y.; Putt, M.E.; Ahima, R.S.; Reilly, M.P. Fractalkine Is a Novel Human Adipochemokine Associated With Type 2 Diabetes. Diabetes 2011, 60, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Zaharieva, E.; Kamenov, Z.; Velikova, T.; Tsakova, A.; El-Darawish, Y.; Okamura, H. Interleukin-18 serum level is elevated in type 2 diabetes and latent autoimmune diabetes. Endocr. Connect. 2018, 7, 179–185. [Google Scholar] [CrossRef]
- Blankenberg, S.; Rupprecht, H.J.; Poirier, O.; Bickel, C.; Smieja, M.; Hafner, G.; Meyer, J.; Cambien, F.; Tiret, L. Plasma Concentrations and Genetic Variation of Matrix Metalloproteinase 9 and Prognosis of Patients With Cardiovascular Disease. Circulation 2003, 107, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Damås, J.K.; Boullier, A.; Waehre, T.; Smith, C.; Sandberg, W.J.; Green, S.; Aukrust, P.; Quehenberger, O. Expression of Fractalkine (CX3CL1) and its Receptor, CX3CR1, Is Elevated in Coronary Artery Disease and Is Reduced During Statin Therapy. Arter. Thromb. Vasc. Biol. 2005, 25, 2567–2572. [Google Scholar] [CrossRef]
- Versteylen, M.O.; Manca, M.; Joosen, I.A.; Schmidt, D.E.; Das, M.; Hofstra, L.; Crijns, H.J.; Biessen, E.A.; Kietselaer, B.L. CC chemokine ligands in patients presenting with stable chest pain: Association with atherosclerosis and future cardiovascular events. Neth. Hear. J. 2016, 24, 722–729. [Google Scholar] [CrossRef]
- Kraaijeveld, A.; de Jager, S.; de Jager, W.; Prakken, B.; McColl, S.; Haspels, I.; Putter, H.; van Berkel, T.; Nagelkerken, L.; Jukema, J.; et al. CC Chemokine Ligand-5 (CCL5/RANTES) and CC Chemokine Ligand-18 (CCL18/PARC) Are Specific Markers of Refractory Unstable Angina Pectoris and Are Transiently Raised During Severe Ischemic Symptoms. Circulation 2007, 116, 1931–1941. [Google Scholar] [CrossRef]
- Stolla, M.; Pelisek, J.; Von Brühl, M.-L.; Schäfer, A.; Barocke, V.; Heider, P.; Lorenz, M.; Tirniceriu, A.; Steinhart, A.; Bauersachs, J.; et al. Fractalkine Is Expressed in Early and Advanced Atherosclerotic Lesions and Supports Monocyte Recruitment via CX3CR1. PLoS ONE 2012, 7, e43572. [Google Scholar] [CrossRef] [PubMed]
- Velagaleti, R.S.; Vasan, R.S. Heart Failure in the Twenty-First Century: Is it a Coronary Artery Disease or Hypertension Problem? Cardiol. Clin. 2007, 25, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Maisel, A.S.; Krishnaswamy, P.; Nowak, R.M.; Mccord, J.; Hollander, J.E.; Duc, P.; Omland, T.; Storrow, A.B.; Abraham, W.T.; Wu, A.H.; et al. Rapid Measurement of B-Type Natriuretic Peptide in the Emergency Diagnosis of Heart Failure. N. Engl. J. Med. 2002, 347, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Li, M.; Zhang, C.; Zhang, Y.; Du, W. Increased levels of inflammatory biomarker CX3CL1 in patients with chronic obstructive pulmonary disease. Cytokine 2020, 126, 154881. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, H.; Hoshino, T.; Takei, S.; Kinoshita, T.; Okamoto, M.; Kawayama, T.; Kato, S.; Iwasaki, H.; Watanabe, K.; Aizawa, H. Interleukin-18 production and pulmonary function in COPD. Eur. Respir. J. 2008, 31, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Sin, D.D.; Miller, B.E.; Duvoix, A.; Man, S.F.P.; Zhang, X.; Silverman, E.K.; Connett, J.E.; Anthonisen, N.A.; Wise, R.; Tashkin, D.; et al. Serum PARC/CCL-18 Concentrations and Health Outcomes in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2011, 183, 1187–1192. [Google Scholar] [CrossRef]
- Gagnat, A.A.; Gjerdevik, M.; Gallefoss, F.; Coxson, H.O.; Gulsvik, A.; Bakke, P. Incidence of non-pulmonary cancer and lung cancer by amount of emphysema and airway wall thickness: A community-based cohort. Eur. Respir. J. 2017, 49, 1601162. [Google Scholar] [CrossRef]
- Maselli, D.J.; Hanania, N.A. Asthma COPD overlap: Impact of associated comorbidities. Pulm. Pharmacol. Ther. 2018, 52, 27–31. [Google Scholar] [CrossRef]
- Izuhara, K.; Conway, S.J.; Moore, B.; Matsumoto, H.; Holweg, C.T.J.; Matthews, J.G.; Arron, J.R. Roles of Periostin in Respiratory Disorders. Am. J. Respir. Crit. Care Med. 2016, 193, 949–956. [Google Scholar] [CrossRef]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein with Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Hong, L.-Z.; Wei, X.-W.; Chen, J.-F.; Shi, Y. Overexpression of periostin predicts poor prognosis in non-small cell lung cancer. Oncol. Lett. 2013, 6, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-H.; Wang, W.; Lin, Y.; Qian, L.-H.; Zhang, X.-W.; Wang, Q.-B.; Yu, L.-K. Diagnostic and prognostic value of serum periostin in patients with non-small cell lung cancer. Oncotarget 2016, 8, 18746–18753. [Google Scholar] [CrossRef] [PubMed]
- Agache, I.; Ciobanu, C.; Agache, C.; Anghel, M. Increased serum IL-17 is an independent risk factor for severe asthma. Respir. Med. 2010, 104, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Pociask, D.A.; McAleer, J.P.; Chan, Y.R.; Alcorn, J.F.; Kreindler, J.L.; Keyser, M.R.; Shapiro, S.D.; Houghton, A.M.; Kolls, J.K.; et al. IL-17RA Is Required for CCL2 Expression, Macrophage Recruitment, and Emphysema in Response to Cigarette Smoke. PLoS ONE 2011, 6, e20333. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Fooladseresht, H.; Minaee, K.; Bazrafshani, M.R.; Khosravimashizi, A.; Nemati, M.; Mohammadizadeh, M.; Mohammadi, M.M.; Ghaderi, A. Higher circulating levels of chemokine CCL22 in patients with breast cancer: Evaluation of the influences of tumor stage and chemokine gene polymorphism. Tumor Biol. 2014, 36, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Prieto, S.; De Chiara, L.; Rodríguez-Girondo, M.; Vázquez-Iglesias, L.; Rodríguez-Berrocal, F.J.; Fernandez-Villar, A.; Botana-Rial, M.I.; De La Cadena, M.P. Highly Sensitive Marker Panel for Guidance in Lung Cancer Rapid Diagnostic Units. Sci. Rep. 2017, 7, srep41151. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hochwald, S.; Deng, S.; Zhu, Y.; Tan, C.; Zhong, Q.; Zhou, Y.; Zhao, H.; Huang, H. Evaluation of EGF, EGFR, and E-cadherin as potential biomarkers for gastrointestinal cancers. Front. Lab. Med. 2017, 1, 135–140. [Google Scholar] [CrossRef]
- Carpaij, O.A.; Muntinghe, F.O.W.; Wagenaar, M.B.; Habing, J.W.; Timens, W.; Kerstjens, H.A.M.; Nawijn, M.; Kunz, L.I.Z.; Hiemstra, P.S.; Tew, G.W.; et al. Serum periostin does not reflect type 2-driven inflammation in COPD. Respir. Res. 2018, 19, 112. [Google Scholar] [CrossRef]
- Kim, V.; Cornwell, W.D.; Oros, M.; Durra, H.; Criner, G.J.; Rogers, T.J. Plasma Chemokine signature correlates with lung goblet cell hyperplasia in smokers with and without chronic obstructive pulmonary disease. BMC Pulm. Med. 2015, 15, 111. [Google Scholar] [CrossRef] [PubMed]
- Loza, M.J.; Watt, R.; Baribaud, F.; Barnathan, E.S.; Rennard, S.I. Systemic inflammatory profile and response to anti-tumor necrosis factor therapy in chronic obstructive pulmonary disease. Respir. Res. 2012, 13, 12. [Google Scholar] [CrossRef]
- Zou, Y.; Chen, X.; Liu, J.; Zhou, D.B.; Kuang, X.; Xiao, J.; Yu, Q.; Lu, X.; Li, W.; Xie, B.; et al. Serum IL-1β and IL-17 levels in patients with COPD: Associations with clinical parameters. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1247–1254. [Google Scholar] [CrossRef]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. The cytokine network in asthma and chronic obstructive pulmonary disease. J. Clin. Investig. 2008, 118, 3546–3556. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, J.; Schild, K.; Jungck, D.; Urban, K.; Müller, K.; Schweda, E.K.H.; Rupp, J.; Koch, A. The T-Helper Cell Type 1 Immune Response to Gram-Negative Bacterial Infections Is Impaired in COPD. Am. J. Respir. Crit. Care Med. 2011, 183, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, J.; Panek, S.; Yanik, S.D.; Jameel, K.J.; Bendella, Z.; Jungck, D.; Bürger, P.; Bülthoff, E.; Struck, B.; Giannakis, N.; et al. The monocyte-dependent immune response to bacteria is suppressed in smoking-induced COPD. J. Mol. Med. 2019, 97, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, J.; Yakin, Y.; Körber, S.; Grensemann, B.; Bendella, Z.; Boyaci, N.; Gallert, W.-J.; Yanik, S.D.; Jungck, D.; Koch, A.; et al. Simvastatin requires activation in accessory cells to modulate T-cell responses in asthma and COPD. Eur. J. Pharmacol. 2016, 788, 294–305. [Google Scholar] [CrossRef] [PubMed]


