 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Samson Oselusi | + 1938 word(s) | 1938 | 2021-07-09 12:11:54 | | | |
| 2 | Conner Chen | Meta information modification | 1938 | 2021-07-12 07:46:01 | | |
Video Upload Options
The growing antimicrobial resistance (AMR) of pathogenic organisms to currently prescribed drugs has resulted in the failure to treat various infections caused by these superbugs. Therefore, to keep pace with the increasing drug resistance, there is a pressing need for novel antimicrobial agents, especially from non-conventional sources. Several natural products (NPs) have been shown to display promising in vitro activities against multidrug-resistant pathogens. Still, only a few of these compounds have been studied as prospective drug candidates. Drug developers are employing different modern strategies to overcome the challenges. These current drug discovery and design strategies can computationally identify potential liabilities and optimize hit compounds to impact desired drug-like properties prior to expensive synthesis and pre-clinical experiments. In addition, it can computationally process a large set of compounds from virtual combinatorial libraries and high-throughput screening to guide rational decision-making in drug discovery and development. This technique of processing large chemical bioactivity data is called cheminformatics.
1. Overview of Cheminformatics
Cheminformatic is a data mining technique that uses computer and information strategies to solve chemical problems by processing raw data into information and information into knowledge [1][2]. Chemical data processing in this context involves working with chemical structures [3]. Therefore, this strategy for drug developers aims to provide better and faster decision-making processes in discovery and lead optimization [1]. Cheminformatics is gaining much acceptance in the field of computational chemistry. It has great potential, especially in the retrieval and extraction of chemical information, database search for compounds, interactive data mining for molecular graphs, and analyses of chemical diversity [1][3][4][5]. It is relevant, particularly in processing hit compounds from virtual and actual high throughput screenings. Cheminformatic processes such as hit profiling (assessing physiochemical properties, molecular descriptors, and drug-likeness) can guide hit prioritization and hit optimization to identify lead compounds (Figure 1), especially from phenotypic screening.
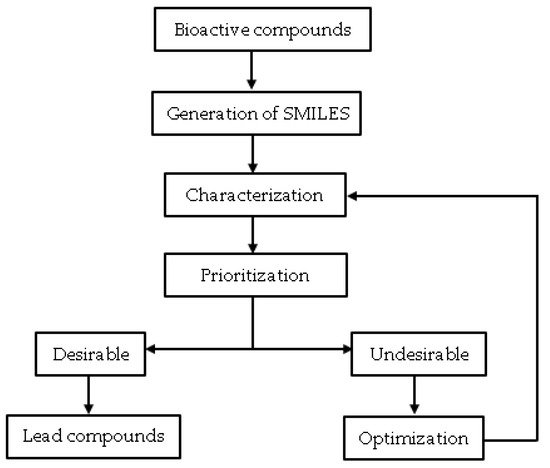
Figure 1. The overall methodology of cheminformatics application in lead discovery.
1.1. Hit Profiling: Physicochemical Properties of NPs
Cheminformatics have played a significant role in the identification of NPs that has the potential to become drug candidates [6]. These techniques are widely used to support traditional wet-lab experiments towards the early identification of drug-like hit, hit-to-lead, and lead optimization processes while improving potency and selectivity. For example, various structural and molecular representations in cheminformatics have proven to help study the molecular complexity and quantify the chemical diversity of a library of compounds. This computational approach has also allowed for profiling, prioritization, and comparison of the molecular descriptors, physicochemical, and pharmacokinetic properties of a group of NPs and others or with those of known drugs [6][7][8][9].
The evaluation of the physicochemical parameters (PP) of potential drug candidates is crucial in drug development, as it assists in the early identification of molecules that may fail at a later stage [10]. The absorption or therapeutic action elicited by a drug depends mainly on the interaction between the various physical and chemical properties of the drug and the targets [11]. Therefore, the physical and chemical properties of any compound are crucial to evaluate the drug-likeness. Furthermore, PP can be manipulated to an optimized condition using computer-aided strategies for a better drug-receptor relationship. The PP that is key to determining the biological activity of any drug candidate has been reviewed [10][11][12][13][14][15], a few of these properties are discussed below.
Molecular Weight (MW)
Molecular weight (MW) is one of the commonly examined physicochemical properties in drug discovery research [16]. This property has been widely studied for its ability to influence various pharmacokinetic properties like absorption, bioavailability, permeation, and elimination, particularly with respect of compounds that are intended for oral administration [17]. MW and few other properties are used in various rule-based drug-likeness filters, such as Lipinski [18] and Ghose [19] to remove undesired compounds from a library. However, antibacterial agents have been reported to deviate from these rules as marketed antibacterial drugs have higher molecular weights than other drugs [20][21]. Furthermore, most marketed antibacterial agents like streptogramins, macrolides, and daptomycin, commonly used against Gram-positive bacteria, possess larger MW than those used against Gram-negative groups [21][22]. However, few Gram-negative bacteria drugs are characterized by substantially high MW. Polymyxin B1 (1203 Da) and azithromycin (749 Da) are examples of these drugs, and they require penetration enhancers to aid their permeability [21].
Partition Coefficient (logP)
The partition coefficient (logP) is the ability of an uncharged molecule to dissolve in a nonhomogeneous two-phase system of lipid and water [23]. It measures the amount of solute that mixes in the water against that which dissolves in a lipophilic portion. The logP is used to evaluate how a molecule travels to the target from the site of administration [11][23]. This implies that the values of logP are significant indicators of the fate of an administered drug in the target organism. A negative logP indicates that the molecule is more hydrophilic, and a positive logP shows that the molecule has a higher affinity for the lipophilic phase.
Similarly, zero logP means that the substance is equally partitioned between the bi-phasic system [23][24]. In order to achieve the desired antimicrobial efficacy, it is important to identify or design compounds with optimum logP that will ensure efficient penetration of the microbes’ cell wall by the natural products. High permeability through microbial cell wall increase efficacy while decreased permeability may give rise to antimicrobial resistance. The ideal logP of active molecules against Gram-negative bacteria was around four, and six, respectively [25].
Hydrogen Bonding
Hydrogen bonding refers to the relationship of an atom of hydrogen from a given compound (known as the donor) and a hydrogen atom from different compounds (known as acceptor), evidenced by bond formation [26][27]. Hydrogen bonds (HBs) are crucial in evaluating the specificity of the binding of a ligand substance to a receptor. The importance of hydrogen bonds in determining the specificity of drug binding has been reported in various studies [28][29][30]. The impact of HBs in the analysis of the quantitative structure-activity relationships (QSAR) model has also been established [11][26]. For example, Kemegne et al. [31] studied the antimicrobial structure-activity relationship of anthraquinones isolated from Vismia laurentii. They reported that hydrogen bond acceptors of the compounds were a determinant of their antimicrobial activity. Furthermore, the addition of a properly positioned HBA side chain (to form an intramolecular HB) may be logical when hydrogen bond donors are required for target activity [32]. Hence, quantifying HBs is vital in identifying and optimizing hit compounds [19].
2. Concept of Drug-Likeness
Drug-likeness is a quantitative concept used to describe molecules that possess functional groups, chemical and physicochemical properties consistent with most of the approved drugs [33][34]. It provides an insight into the early identification of chemical compounds that are “most likely to succeed” in the drug development venture. A commonly used approach for estimating the drug-likeness of a given molecule is to screen against acceptable boundaries of some fundamental molecular properties. An example of this strategy is the famous “Rule of Five’’ developed by Lipinski et al. [18]. Ghose [19] and Veber’s rule [35], among many other property-based rules, have also been used in various studies to determine drug-likeness [36]. The question is whether the application of these drug-likeness estimation strategies to natural products is a comparison of apples with oranges? Natural products, chemical entities produced by living organisms, tend to break these established drug-likeness rules obtained from synthetic chemical libraries. The concept of natural product-likeness has been reported to have the potential to open new opportunities for drug discovery from natural compounds while neglected by the drug-likeness rule [34].
2.1. Lipinski’s Rule of Five (Ro5)
The Ro5 is a collection of some important PP that needs to be prioritized in determining the success of orally administered drugs [11][37][38]. There are a likelihood for poor absorption and permeability for drug candidates whose logP, hydrogen bond donors (HBDs), hydrogen bond acceptors (HBAs), and molecular weight (MW) is above 5, 5, 10, and 500, respectively [36][37][38][39]. The digit 5 in Ro5 indicates the limit of the parameters, multiples of 5 [11]. This strategy aims to use a drug-likeness filter to identify for quickly; removal or optimization of poor pharmacokinetic compounds at an earlier stage of drug discovery [36][38][39].
Several authors have explained successful cases where Ro5 has been employed to evaluate the drug-likeness of hundreds and thousands of NPs [38][40][41]. Zhang and Wilkinson [42] also reported that about two-thirds of the FDA-approved drugs are administered orally and passed the Ro5. However, some drawbacks have been identified with the use of Lipinski’s rule. For example, approved drugs, such as atorvastatin, bromocriptine, and everolimus are notable violators of the Ro5 [43][44]. Similarly, Zhang and Wilkinson [42] have reported that 20% of all orally administered drugs failed at least one of the parameters of Lipinski’s rule.
2.2. Pharmacokinetics and Toxicity Parameters
Pharmacokinetic descriptors such as absorption, distribution, metabolism, and excretion (ADME), and toxicity (T) are commonly used properties for profiling or predicting the fate of many drug candidates after clinical administration [45]. The concept of investigating the ADMET is of interest in early drug discovery given that over 70% of clinical failures have been connected to these properties [46][47]. In addition to potency, a successful drug candidate is expected to have favorable ADMET properties [45][47].
The use of in silico methods in determining these parameters has significantly contributed to recent advancements in discovery and development [11]. For instance, ADMET profiling has been used in various studies to identify lead compounds [48][49]. In addition, the assessment of the ADMET properties for potential drug candidates could guide computational chemists towards an effective structure-activity relationship (SAR) based optimization [47][50].
3. Hit-Prioritization Using the Quantitative Estimate of Drug-Likeness
To address the constraints of the rule-based filtering of compounds, Bickerton et al. [33] developed a quantitative estimate of drug-likeness (QED) by combining the desirability of key physicochemical properties (such as molecular weight, polarity, numbers of hydrogen bond acceptors, and donors, lipophilicity, and the number of structural alerts) , which impacts the likelihood of attrition [36][51][52]. The QED is a flexible and continuous metric score whose value ranges between 0 and 1. A score of 1 in this context describes any chemical compound with all its physicochemical properties within the space of an ideal oral drug-like profile, while a score of 0 describes a compound with undesired properties [24][52][53]
The concept of QED has been used in various studies to prioritize large compound sets and their drug targets. For example, Egieyeh et al. [53] conducted cheminformatic profiling of 1040 NPs with anti-plasmodial activity. They generated a list of compounds that can be prioritized in the development of anti-malarial drugs. Similarly, a collection of more than 100 active compounds against methicillin-resistant Staphylococcus aureus (MRSA) was also prioritized for anti-MRSA drug development in a recent study [24]. Kim and Lee [54] also screen chemical compounds obtained from a Chinese medicinal plant. They used the QED concept as one of the approaches to profile 475 active compounds for drug-likeness and oral bioavailability. In all these studies, QED has been described as a more reliable method to estimate drug-likeness than the rule-based approaches [36][52].
4. Hit Optimization after Hit Profiling
The aim of structurally optimized hit compounds is to enhance the development of potential drug candidates. In silico cheminformatic tools can help enhance the physicochemical and pharmacokinetic properties of hit compounds. This is achieved by selectively modifying the structure of such compounds [55][56][57]. In general, this strategy also tends to optimize the compounds toward reducing toxicity, improving ADME properties, and synthetic accessibility while maintaining the desired potency [55][56].
Structural optimization in drug design can be carried out through a combination of different approaches [57]. The simplest of these strategies is the direct chemical modification of functional groups through isosteric replacement, addition, and alteration of the ring systems [58]. This strategy is based on the chemical similarity principle, which states that chemically similar structures will have similar bioactivity. In a recent study [24], random replacement of the functional group was performed on two chemical compounds, α-viniferin and aminoethyl-chitosan, which showed good anti-MRSA activity but a low desirability score. This led to the identification of two compounds with a significantly improved properties and a better desirability score.
References
- Gillet, V.J. Applications of Chemoinformatics in Drug Discovery 2. 2 Computer Representation of Chemical Structures; Wiley: Hoboken, NJ, USA, 2019.
- Egieyeh, S.; Malan, S.F.; Christoffels, A. Cheminformatics techniques in antimalarial drug discovery and development from natural products 1: Basic concepts. Phys. Sci. Rev. 2019, 4, 1–11.
- Xu, J.; Hagler, A. Chemoinformatics and drug discovery. Molecules 2002, 7, 566–600.
- Jónsdóttir, S.Ó.; Jørgensen, F.S.; Brunak, S. Structural bioinformatics Prediction methods and databases within chemoinformatics: Emphasis on drugs and drug candidates. Bioinformatics 2005, 21, 2145–2160.
- Medina-Franco, J.L. Advances in computational approaches for drug discovery based on natural products. Rev. Latinoam. Química 2013, 41, 95–110.
- Chen, Y.; Kirchmair, J. Cheminformatics in Natural Product-based Drug Discovery. Mol. Inform. 2020, 39, 2000171.
- Wetzel, S.; Schuffenhauer, A.; Roggo, S.; Ertl, P.; Waldmann, H. Cheminformatic analysis of natural products and their chemical space. Chim. Int. J. Chem. 2007, 61, 355–360.
- Willett, P. From chemical documentation to chemoinformatics: 50 years of chemical information science. J. Inf. Sci. 2008, 34, 477–499.
- Medina-Franco, J.L.; Saldívar-González, F.I. Cheminformatics to characterize pharmacologically active natural products. Biomolecules 2020, 10, 1566.
- Wenlock, M.C.; Barton, P. In silico physicochemical parameter predictions. Mol. Pharm. 2013, 10, 1224–1235.
- Chandrasekaran, B.; Abed, S.N.; Al-Attraqchi, O.; Kuche, K.; Tekade, R.K. Computer-aided prediction of pharmacokinetic (ADMET) properties. In Dosage Form Design Parameters; Elsevier: Amsterdam, The Netherlands, 2018; pp. 731–755.
- Gleeson, M.P.; Leeson, D.; Waterbeemd, H.A.N.V.A.N.D.E. Physicochemical Properties and Compound Quality. In The Handbook of Medicinal Chemistr; The Royal Society of Chemistry: London, UK, 2015; pp. 1–31. ISBN 9781782621836.
- Schneider, G. Prediction of drug-like properties. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013.
- Vallianatou, T.; Giaginis, C.; Tsantili-Kakoulidou, A. The impact of physicochemical and molecular properties in drug design: Navigation in the “drug-like” chemical space. In GeNeDis 2014; Springer: Berlin/Heidelberg, Germany, 2015; pp. 187–194.
- Lobell, M.; Hendrix, M.; Hinzen, B.; Keldenich, J.; Meier, H.; Schmeck, C.; Schohe-Loop, R.; Wunberg, T.; Hillisch, A. In silico ADMET traffic lights as a tool for the prioritization of HTS hits. ChemMedChem 2006, 1, 1229–1236.
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456.
- Lagorce, D.; Douguet, D.; Miteva, M.A.; Villoutreix, B.O. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep. 2017, 7, 1–15.
- Lipinski, C. Physicochemical properties and the discovery of orally active drugs: Technical and people issues. In Proceedings of the In Molecular Informatics: Confronting Complexity, Beilstein-Institute Workshop, Bozen, Italy, 13–16 May 2002; pp. 59–78.
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68.
- Ebejer, J.P.; Charlton, M.H.; Finn, P.W. Are the physicochemical properties of antibacterial compounds really different from other drugs ? J. Cheminform. 2016, 8, 1–9.
- O’Shea, R.O.; Moser, H.E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51, 2871–2878.
- Konaklieva, M.I. Addressing Antimicrobial Resistance through New Medicinal and Synthetic Chemistry Strategies. SLAS Discov. Adv. Life Sci. R&D 2019, 24, 419–439.
- Bhal, S.K. LogP—Making Sense of the Value; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2007; pp. 1–4.
- Oselusi, S.O.; Egieyeh, S.A.; Christoffels, A. Cheminformatic Profiling and Hit Prioritization of Natural Products with Activities against Methicillin-Resistant Staphylococcus aureus, (Forthcoming). 2021.
- Ropponen, H.K.; Richter, R.; Hirsch, A.K.; Lehr, C.M. Mastering the Gram-Negative Bacterial Barrier–Chemical Approaches to Increase Bacterial Bioavailability of Antibiotics. Adv. Drug Deliv. Rev. 2021, 172, 339–360.
- Schwöbel, J.A.H.; Ebert, R.; Kühne, R.; Schüürmann, G. Prediction models for the Abraham hydrogen bond donor strength: Comparison of semi-empirical, ab initio, and DFT methods. J. Phys. Org. Chem. 2011, 24, 1072–1080.
- Yunta, M.J. It is important to compute intramolecular hydrogen bonding in drug design. Am. J. Model. Optim. 2017, 5, 24–57.
- Wade, R.C.; Goodford, P.J. The role of hydrogen-bonds in drug binding. Prog. Clin. Biol. Res. 1989, 289, 433–444.
- Zhao, H.; Huang, D. Hydrogen bonding penalty upon ligand binding. PLoS ONE 2011, 6, e19923.
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240.
- Lacret, R.; Oves-Costales, D.; Gómez, C.; Díaz, C.; De la Cruz, M.; Pérez-Victoria, I.; Vicente, F.; Genilloud, O.; Reyes, F. New ikarugamycin derivatives with antifungal and antibacterial properties from Streptomyces zhaozhouensis. Mar. Drugs 2015, 13, 128–140.
- Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608.
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98.
- Ntie-Kang, F.; Nyongbela, K.D.; Ayimele, G.A.; Shekfeh, S. “Drug-likeness” properties of natural compounds. In Fundamental Concepts; De Gruyter: Berlin, Germany, 2020; pp. 81–102. ISBN 3110579359.
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623.
- Mignani, S.; Rodrigues, J.; Tomas, H.; Jalal, R.; Singh, P.P.; Majoral, J.P.; Vishwakarma, R.A. Present drug-likeness filters in medicinal chemistry during the hit and lead optimization process: How far can they be simplified ? Drug Discov. Today 2018, 23, 605–615.
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341.
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10.
- Krämer, S.D.; Aschmann, H.E.; Hatibovic, M.; Hermann, K.F.; Neuhaus, C.S.; Brunner, C.; Belli, S. When barriers ignore the “rule-of-five”. Adv. Drug Deliv. Rev. 2016, 101, 62–74.
- Boufridi, A.; Quinn, R.J. Harnessing the properties of natural products. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 451–470.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Paul, J.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17.
- Zhang, M.-Q.; Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007, 18, 478–488.
- Abad-Zapatero, C. A Sorcerer’s apprentice and the rule of five: From rule-of-thumb to commandment and beyond. Drug Discov. Today 2007, 12, 995–997.
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98.
- Vrbanac, J.; Slauter, R. ADME in drug discovery. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development; Elsevier: Amsterdam, The Netherlands, 2017; pp. 39–67.
- Sharma, A.; Saini, R. Pharmacokinetic profiling of anticancer phytocompounds using computational approach. Phytochem. Anal. 2018, 29, 559–568.
- Wang, J.; Urban, L. The impact of early ADME profiling on drug discovery and development strategy. DDW Drug Discov. World 2004, 5, 73–86.
- Skariyachan, S.; Krishnan, R.S.; Siddapa, S.B.; Salian, C.; Bora, P.; Sebastian, D. Computer aided screening and evaluation of herbal therapeutics against MRSA infections. Bioinformation 2011, 7, 222.
- Qureshi, S.I.; Chaudhari, H.K. Design, synthesis, in-silico studies and biological screening of quinazolinone analogues as potential antibacterial agents against MRSA. Bioorg. Med. Chem. 2019, 27, 2676–2688.
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20.
- Yusof, I.; Segall, M.D. Considering the impact drug-like properties have on the chance of success. Drug Discov. Today 2013, 18, 659–666.
- Kirkpatrick, P. Shades of chemical beauty. Nat. Rev. Drug Discov. 2012, 11, 107.
- Egieyeh, S.A.; Syce, J.; Malan, S.F.; Christoffels, A. Prioritization of anti–malarial hits from nature: Chemo–informatic profiling of natural products with in vitro antiplasmodial activities and currently registered anti–malarial drugs. Malar. J. 2016, 15, 1–23.
- Kim, S.-K.; Lee, S. Drug-likeness and Oral bioavailability for Chemical Compounds of Medicinal Materials Constituting Oryeong-san. Korea J. Herbol. 2018, 33, 19–37.
- Guo, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136.
- Chen, Y.; Zhao, J.; Qiu, Y.; Yuan, H.; Khan, S.I.; Hussain, N.; Choudhary, M.I.; Zeng, F.; Guo, D.; Khan, I.A.; et al. Fitoterapia Prenylated fl avonoids from the stems and roots of Tripterygium wilfordii. Fitoterapia 2017, 119, 64–68.
- Xiao, Z.; Morris-Natschke, S.L.; Lee, K.H. Strategies for the Optimization of Natural Leads to Anticancer Drugs or Drug Candidates. Med. Res. Rev. 2016, 36, 32–91.
- Harrold, M.W.; Zavod, R.M.; Harrold, M.W.; Zavod, R.M. Functional group characteristics and roles. In Basic Concepts in Medical Chemistry; Harrold, M.W., Zavod, R.M., Eds.; American Society of Health-System Pharmacists: Bethesda, MD, USA, 2013; pp. 15–50.

