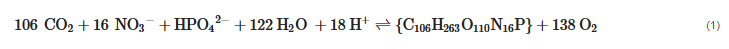The forward and backward reaction in Equation (1) indicates the photosynthesis and the respiration, respectively. The significance of Equation (1), in addition to the photosynthesis and natural respiration, is the involved chemical cycles of carbon, nitrogen, and phosphorus. This further implies that the reclamation of trace nutrients is an additional benefit to biotic
CO2
conversion. By contrast, artificial CO2 conversion requires energy input since CO2 has no heat value. Recent progress in catalysis greatly reduces the energy requirement in artificial CO2 reduction (CO2R).
There are excellent reviews on the mechanistic aspects and effectiveness of CO2R catalysts [
2,
3,
4,
5,
6]. Artificial CO2R can be achieved by processes, such as electrochemical [
7,
8,
9], photochemical [
10,
11,
12], and photoelectrochemical (PEC), by which electrons are transferred to CO
2 molecules to increase their energy content [
13,
14]. Electrochemical CO2R is an electron transfer reaction between the cathode and the adsorbed CO
2. Photochemical CO2R relies on the transfer of photogenerated electrons in the LUMO (the lowest unoccupied molecular orbital) of the catalyst to the HUMO (the highest occupied molecular orbital) of adsorbed CO
2. In photoelectrochemical (PEC) CO2R, the photogenerated electrons are transported to the cathode in the presence of a bias electric field. The external electric field effectively suppresses the recombination loss of photogenerated charge carriers and enhances CO2R efficiency.
In all modes of CO2R systems, catalysts (in the form of photosensitive or electric conductive) are needed to facilitate the CO2 reduction reactions. Therefore, the stability and surface chemistry of catalysts (electrodes) become important operation parameters. For instance, it is suggested that a thin γ−Al2O3
overlayer could effectively stabilize the Faradaic efficiency and the partial current density of the SnO
2 catalyst in an electrochemical CO2R reaction [
8]. Chang et al. (2016) have reported that photogenerated holes, not electrons, are the primary cause of instability of Cu
2O catalysts, which require Cu
2O to be operated as dark cathodes [
13]. High-CO-affinity electrocatalysts (i.e., Cr, Mn, and Fe-N-C) exhibited high carbon monoxide (CO) Faradaic efficiency. The pyridinic and hydrogenated (pyrrolic) nitrogen moieties of the carbonaceous support are active sites for CO
2 adsorption [
9]. Accordingly, a relatively basic surface (such as the presence of
γ−Al2O3
overlayer and pyridinic modification) would have a positive effect on CO2R efficiency enhancement, likely through increasing the affinity of the catalyst surface toward CO
2 adsorption. Defects in a catalyst would introduce coordinately unsaturated sites (i.e., active sites for molecular chemisorption) and provide spatially supply channels for energy and electron transfers in photochemical CO2R [
10]. Note that among all available CO2R processes, electrochemical CO2R is probably the simplest; therefore, it is the easiest and most sustainable (relative to chemical reduction) for operations and scaling-up.
CO2R involves the consecutive transfer of one electron or hydrogen from the catalyst to CO
2 and intermediates, which gradually reduces the carbon oxidation number stepwise. Only intermediates that possess even oxidation numbers are thermodynamically stable. This is why formic acid (FA, HCOOH), carbon monoxide (CO), formaldehyde (HCOH), methanol (CH
3OH), and methane (CH
4) are always found in CO2R reactions. This review focuses on recent advances of CO2R systems, both biotic and abiotic processes, and as far as the broad view of CO
2 conversion is concerned, three significant implications are noted [
15].
Mitigating greenhouse gas effect: Electrocatalytic CO2R is usually conducted with high-purity CO
2 based on thermodynamics considerations. This means that additional electricity is required for concentrating CO
2 feedstock; however, the majority of electricity in modern society is produced through fossil fuels. Adopting electrocatalytic CO2R for the mitigation of the greenhouse effect would instead increase atmospheric CO
2 concentration [
2,
16]. Therefore, increasing the deployment of renewable energy would be equally important in addition to the efficiency improvement of electrocatalytic CO2R from the prospect of greenhouse gas mitigation.
Electrochemical conversion of CO
2 to fuels: In order to achieve a carbon neutral footprint through electrocatalytic CO2R, produced hydrocarbons should not be fed to the combustion engines directly. Instead, they should be feed for fuel cells [
15]. Again, increasing the efficiency of fuel cells for feeding CO
2-derived methanol [
17,
18] or formic acid [
19,
20] is another issue of concern. Selectivity of CO2R catalysts is crucial too as it minimizes the carbon footprint required for subsequent separation and concentration of hydrocarbons produced from electrocatalytic CO2R [
21].
Electrochemical conversion of CO
2 to a building block: Electrocatalytic CO2R to carbonaceous fuels usually leads to a positive carbon footprint as mentioned above. However, a carbon neutral footprint could be possibly reached by converting CO
2 to a building block as an alternative. This is because CO
2 is a C1 building block that requires no extra energy for its production [
22]. For example, carbon monoxide produced by electrocatalytic CO2R could be directly used as the source for producing phosgene [
23,
24]. Similarly, as-obtained methanol and formic acid could be feedstocks for reversible chemical hydrogen storage and other applications [
5,
25].
Apparently, converting CO
2 to a building block for chemicals production could entice the pursuant of electrocatalytic CO2R. Indeed, all conceptual designs of an economically affordable CO2R business for achieving a negative carbon footprint have assumed that the CO2R catalysts could exhibit excellent reactivity and selectivity. For instance, Gai et al. (2016) performed a conceptual design of methanol production from CO
2 at an industrial scale using ASPEN Plus
® (Bedford, MA, USA) in which CO and H
2 are produced from the electrolysis of CO
2 and H
2O [
26]. They found that when CO
2 conversion is less than 42%, the optimal methanol synthesis route is CO hydrogenation. Importantly, the achievement of a near zero carbon emission power plant is strongly built on the assumption that CO is the only intermediate and no additional energy is required for CO isolation [
26]. Sun et al. (2019) have developed a 20 MWth solar–wind biodistributed energy system for simultaneously biomass cascade utilization, water resource conservation, waste heat recovery, and CO
2 mitigation for hydrogen, formic acid, and grapheme production [
27]. Again, in their framework, the energy efficiency is vulnerable to the compromised selectivity of electrocatalytic CO2R. Based on the above considerations, it is clear that the success of a CO2R industry strongly relies on multidiscipline cooperation and that technology is part of this. Additional bonuses such as creating jobs, building blocks for chemicals, and carbon right trading would make CO2R more sustainable. Integrating the knowledge of biotic and abiotic CO2R is another useful approach. A good example is the microbial electrosynthesis system (MES), in which the microbial is responsible for biotic CO2R, while engineering the electrode (abiotic CO2R) further improves the overall CO2R efficiency. Accordingly, recent advances in abiotic CO2R and biotic CO2R will be reviewed herein. Having a deep insight into the scientific and technological aspects of both abiotic and biotic CO2R would advance the design of efficient catalysts and the microalgae farming system. We first focus on the technology aspect of abiotic CO2R by discussing the reactivity and selectivity of CO2R catalysts and reaction mechanisms in both water and gas phases. The effect of the surface chemistry of synthesized catalysts on the reactivity and selectivity of CO2R will be addressed. This will be particularly beneficial to the rational design of high-efficient catalysts for CO2R conversion. CO2R through microalgae abstraction of CO
2 is highly influenced by the bioactivity of selected microalgae. Separation and purification of various value-added chemicals obtained is another issue of concern, and further refinery of algal biomass is also included in this review. Additional considerations such as the involvement of other stakeholders that allow CO2R to be more sustainable are also discussed.
2. The Chemistry of Abiotic CO2R
2.1. Abiotic CO2R in Water Phase
CO2R can occur in the water or gas phase. In the former system, carbonate species, namely, H
2CO
3*, HCO
3−, CO
32−, are reduced. In the latter, gaseous CO
2 is reacted with electron donors over catalysts and C1 or C2 compounds such as CO, formate, methanol, and oxalate are major products. Undoubtedly, the usage of rare and precious metals, such as Re and Pd, always leads to the highest CO2R efficiency [
4]. The application of transition metals such as Fe, Mn, and Ni has received much attention recently because of material abundance and economical affordability [
33,
34].
2.1.1. Effect of Cu Surface Chemistry on Abiotic CO2R
Buckley et al. (2019) have studied the structure-reactivity relationships of electrocatalytic CO2R on modified Cu cathode surfaces [
35]. The Cu cathode is first modified with long chain hydrocarbons so as to render the Cu surface hydrophobic. Modified electrodes are used to study the CO2R reaction in CO
2-saturated KHCO
3 (0.05 M) solution [
36]. The Faradaic efficiency (η
F) of each species is calculated by Equation (2):
where F is the faraday constant (96,485 C-mol−1), V is the volume of electrolyte (specifically the catholyte), C is the concentration of carbonate species (M), and Q is the total charge passing through the cathode during electrocatalysis CO2R. n is the number of electron transfer in the CO2R process. For example, n = 2 for CO2 reduction to CO or HCOOH (FA). The partial current density (ji) of certain species is the product of the Faradaic efficiency and total current density (jtotal):

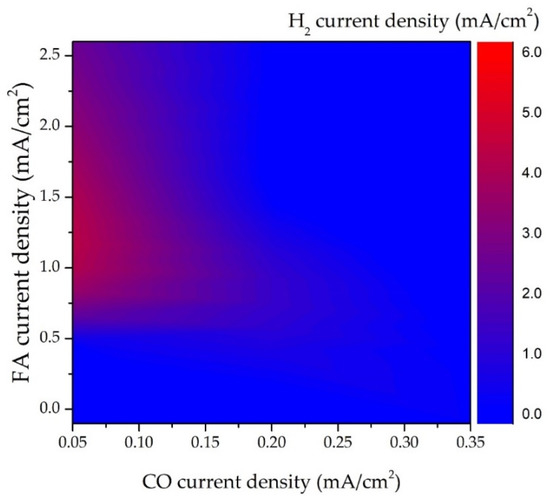
Figure 1 shows the replotted contour image of the current density of FA, CO, and H
2 production in electrocatalytic CO2R over the modified Cu cathode [
35]. Two interesting features are noted in
Figure 1. First, FA is the major CO2R species on Cu-based cathodes as its value is about tenfold higher than that of CO. Second, the hot zone (the red area) of the hydrogen current occurs at the left-handed side of the figure that corresponds to the region with the lowest CO current. This clearly indicates that electrocatalytic hydrogen production profoundly competes with CO evolution. Importantly, the hot zone of hydrogen evolution appears in the region with a moderate FA current (0.5~2.5 mA/cm
2). This means that FA formation is less affected by hydrogen evolution than that of the CO formation [
13]. It must be mentioned that increasing operation potential inevitably leads to high H
2 yield (
Figure 2) [
37]. Due to thermodynamics restrictions, FA and CO evolution in CO2R is suggested to be carried out at a relatively low potential condition.
Figure 1. Current density of FA, CO, and H
2 production in electrocatalytic CO2R over modified Cu electrode. Replotted data from Buckley et al. (2019) [
35].
Figure 2. Faradaic efficiency of CO, FA, and H
2 production in electrocatalytic CO2R over boron-doped diamond (BDD) cathodes as a function of operation potential. Replotted data from Tomisaki et al. (2019) [
37].
Engineering electrochemical properties of cathode materials enhances the selectivity. Designing an efficient catalyst for CO2R is a highly technical challenge due to the strong completion from the hydrogen evolution reaction (HER) [
3]. Factors such as catalytic reactivity, product selectivity, Faradaic efficiency, catalytic stability, and reduction mechanisms are crucial to controlling the efficiency of CO2R [
38]. A systematical evaluation of the effect of catalyst structure on reaction selectivity is hence highly desirable [
4]. Taking the electrocatalytic CO2R molecular system as an example, the energy required to dissociate an M–H bond to form a hydride is the key parameter in CO2R selectivity [
2,
4,
39,
40].
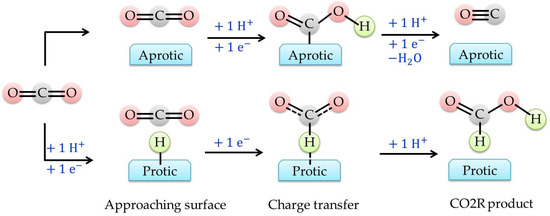
Figure 3 shows pathways regulating the transfer of either two protons (for CO evolution) or two electrons (for formic acid formation) and in both cases hydrogen evolution is always the major competitive side reaction.
Figure 3. Illustration of CO2R pathways occurring at the protic and aprotic surface. Rearranged from Buckley et al. (2019) [
35].
In this framework, the preferential interaction between the catalytic metal center and CO
2 over protons is responsible for the selectivity for CO evolution. While the moderate hydricity facilitates CO
2 insertion into M–H bonds for FA production, strong hydride donors catalyze H
2 formation [
2,
4,
39,
40]. It has been suggested that catalytic activity requires the presence of a weakly coordinating solvent molecule that can readily become dissociated during the catalytic cycle as to provide a vacant coordination site for water binding and assisting C−O bond cleavage [
4]. Generally, H
2 and FA formations are favorable reactions in aqueous solutions [
39]. In fact, by plotting the hydricity as a function of individual free energy for the one-electron reduction of the parent species, a linear correlation appears indicating high FA selectivity over CO formation. Importantly, this correlation extends over a wide range of metals, ligand architectures, structural geometries, and overall charge of the metal hydride [
40]. High overpotential is always found in CO2R [
41]. Despite hydrogen evolution always being a competing reaction in CO2R, it is still worthy of scientific investigation on the hydrogenation of CO
2 to FA and dehydrogenation of FA as a practical hydrogen storage pathway [
5]. FA formation essentially increases the density of hydrogen gas [
6]. This opens a practical alternative, such as the direct formic acid fuel cells (DFAFC) [
42]. Major heterogeneous metal catalysts, such as In, Sn, Hg, and Pb exhibit high FA selectivity [
5,
6,
41,
42]. Electrochemical CO2R on polycrystalline Sn surfaces exhibits high FA selectivity too. Formation of *OCHO at Sn surfaces is the key intermediate for FA production due to optimal *OCHO binding energy. The results suggest that oxygen-bound intermediates are critical to understanding the mechanism of CO
2 reduction to HCOO– on metal surfaces [
43].
Cu is known to have relatively low CO selectivity because the CO produced is further reduced to several multi-carbon oxygenates (i.e., ethanol, acetate, and n-propanol) [
41]. Specifically, sulfur-modified copper catalysts (Cu–S) exhibit positive correlation between particle size and selectivity toward FA evolution [
44]. Nanostructured porous dendritic Cu-based catalysts show stable and selective conversion of CO
2 into FA at high current density with low overpotential [
45]. The relatively low CO selectivity on Cu surfaces results from consecutive CO electroreduction activity [
46]. That is, in CO
2-saturated aqueous solutions, polycrystalline Cu catalysts produce a mixture of compounds. Indeed, H
2 evolution is dominated at low overpotential, CO and FA formation mainly occurs at high overpotential, while hydrocarbons, ethanol, acetate, and n-propanol formation happen at the most extreme overpotentials [
47,
48]. In a CO
2 free environment, CO is reduced to hydrocarbons and multi-carbon oxygenates over the Cu catalyst [
49,
50]. Interestingly, oxide-derived Cu (Cu catalysts prepared by reducing Cu
2O) shows much higher H
2 selectivity than polycrystalline Cu [
51]. Similarly, aqueous electrochemical CO reduction to C2 products by face-to-face coordinated thiol-terminated metalloporphyrins on copper electrodes exhibits 83% Faradaic efficiency and 1.34 mA/cm
2 current density at −0.40 V vs. RHE. This is a significant improvement in both selectivity and activity by one order of magnitude over parent copper surfaces or copper functionalized with porphyrins in an edge-on orientation [
52]. In a similar system, oxide-derived copper (OD-Cu) electrodes exhibit a high CO reduction performance by producing ethanol and acetate with >50 % Faradaic efficiency at −0.3 V vs. RHE [
53].
In a short summary, the selectivity of abiotic CO2R in the aqueous phase is highly sensitive to the surface chemistry of Cu-family catalysts. CO2R occurring at the aprotic surface tends to yield CO as the major product. By contrast, CO2R happening at the protic surface is prone to produce hydrocarbons and multi-carbon oxygenates as the major products. In this case, the selectivity is strongly affected by the involved reaction pathway. In the former case, the reduction is achieved through the charge transfer, while in the latter case the hydride transfer is mainly responsible for the CO2R. The adsorption affinity between reduced intermediates and Cu-family catalysts is another critical factor regulating the CO2R selectivity. High adsorption affinity slows the desorption of the reduced intermediates, which enables their consecutive reduction. This explains that the hydrocarbons and multi-carbon oxygenates as the major products are found in this case. It is thus concluded that increasing CO2R selectivity could be achieved through the modification of surface hydrophobicity and adsorption affinity.
2.1.2. Effect of Surface Chemistry of Non Cu-Family Catalysts on Abiotic CO2R
In the iron-based CO2R, introducing the extra elemental Fe plate profoundly decreases the overpotential of the microbial electrosynthesis system (MES) [
54]. In this MES system, all produced CO
2 is reduced to formate at the cathode and vast hydrogen is produced during the digestion of waste activated sludge. This is attributed to the high selectivity toward formic acid evolution over CO and methane production to the reduction of H
+ at the cathode due to the slow methanogensis in Fe-C MES [
54]. In the case of photocatalytic CO2R, enhancing sunlight conversion efficiency is always accompanied with improving CO2R selectivity [
55]. Similar to the strategy adopted in the dye-sensitized photoelectrochemical cells [
56], increasing selectivity in FA formation is usually achieved through coordinating active metals with covalent organic frameworks. For example, the columnar orientation COFs (covalent organic frameworks) provides a high-efficient charge carrier transport through the ordered π-electronic pathway, which improves electron transfer from COF to metal moiety and thus increases the reactivity [
55]. The results of density functional theory computations further reveal that COFs decorated with electron-donating substituents favor CO
2 reduction by decreasing the hydricity of the Rh–H bond. This results in a lower hydride transfer barrier toward formic acid production [
57] because the selectivity toward CO or HCOOH production is dependent on the coordination environment of the metal ion being capable of cleaving the C–O bond in the metal–CO
2H intermediate [
55]. Specifically, an electron-rich coordination environment breaks the C−O bond to form CO, whereas an electron-deficiency coordination environment tends to enhance the C−O bonding thereby enhancing FA formation [
58]. That is, if the center metal in covalent-organic frameworks (COFs) is a strong π-donor, such as Co(II), it usually tends to promote CO evolution. By contrast, a weak π-donor, such as Zn(II), favors HCOOH production [
55]. A catalyst exhibiting low adsorption energy for HCOO* (i.e., a large energy difference between the two adsorbed CO
2 reduction intermediates, namely HCOO* and COOH* and large H* adsorption energy) would have high FA selectivity [
59]. Ajmal et al. (2019) have studied the selectivity of the CO
2 reduction reaction over bimetallic CuZn alloy catalysts and reported that the Faradaic efficiency and partial current density of FA on Cu
0.5Zn
0.5 (equal molar ratio of Cu and Zn) are enhanced by nearly 4 and 5 times, respectively, that of Cu foil [
60]. The high selectivity of the CuZn bimetallic alloy catalyst is originated from the synergistic effect of Cu and Zn. In this case, the Zn (a weak π-donor) is likely to create a large energy difference for the adsorption of two CO
2 reduction intermediates, namely, HCOO* and COOH*. An et al. (2019) have studied CO
2 reduction over Sn/SnOx catalysts and reported a maximum FA Faradaic efficiency of 89% at −1.7 V (vs. Ag/AgCl) in a 0.1 M CO
2-saturated KHCO
3 solution [
61]. The authors further concluded that Sn(IV) and Sn(II) species are mainly responsible for controlling the overpotential and suppressing H
2 evolution toward improved FA selectivity. Chatterjee et al. (2019) have reported that nanoporous Pd-based alloys (np-PdX, X = Co, Ni, Cu, and Ag) exhibit FA selectivity following the order: np-PdAg > np-PdCu > Pd/C > np-PdNi > np-PdCo [
62]. They have concluded that the composition-dependent behavior was governed by CO adsorption strength associated with the presence of transition metal alloying components near the Pd-skin surface and a composition-dependent change in the near surface H-sorption capacity. Interestingly, the free-standing np-PdCo and np-PdNi catalysts are able to sustain a high formate partial current density (>20 mA-cm
−2) with high CO poisoning tolerance while exhibiting insignificant loss of the active area [
62]. This further highlights the importance of durability and resistance of catalysts against CO poisoning during CO2R.
The results of computational hydrogen electrode model simulation reveal a striking similarity in CO2R electrocatalytic activity for the Cu
3 vs. Cu
5 and Cu
4 vs. Cu
6 size-selected clusters [
63]. The rate-limiting potential of Cu
4 and Cu
6 clusters in CO2R is the proton-electron (H
+ + e
−) transfer to CO* (species adsorbed on clusters) to form CHO*, which is also the rate-limiting step on Cu surfaces. On the other hand, with respect to Cu
3 and Cu
5 clusters, removing OH* from the cluster surface (OH*
→S+OH)is the rate-limiting step in CO2R [
63]. The above simulation unambiguously implies the role of surface defects, in addition to bulk electrocatalysts, in regulating CO2R reaction pathways. Indeed, the electrolysis of CO
2 on 4-aminomethylbenzene-modified Pb electrodes exhibits a current density as high as 24.0 mA/cm
2 (at −1.29 V vs. RHE) and a FA Faradaic efficiency greater than 80% [
28]. Pt-based alloys having high-index facets generally show high specific catalytic activity over those having low-index facets [
64]. Exposing the high-index facets of nanosized particles is promising to enhance Pt utilization and at the same time enriches crystalline defects in the CO2R catalyst [
64]. Similarly, Pan et al. (2019) have reported that N,S-codoped carbon catalysts exhibit 92% CO Faradaic efficiency and CO current density of 2.63 mA/cm
2 at a low overpotential of 0.49 V versus RHE [
65]. Incorporating S in N-doped carbon introduces a high population of activate pyridinic N sites, which significantly decreases the free energy barrier for the formation of intermediate *COOH thereby enhancing CO adsorption toward high CO selectivity [
65]. The high CO selectivity of the Pd
85Cu
15 catalyst is attributed to the presence of a larger number of low-coordination Cu sites than active monometallic Pd sites on the catalyst [
66]. Accordingly, manipulating the size and chemical composition of bimetallic nanoparticles is critical to the selectivity of CO2R [
66]. Results of density functional theory calculation indicate that high reactivity and selectivity are the outcome of defects that stabilize the *OCHO intermediate [
67]. Surface modification of the Cu catalyst with protic, hydrophilic, and cationic hydrophobic species results in increasing the selectivity of H
2, FA, and CO, respectively [
35].
Table 1 summarizes the performance of various CO2R processes. Note that Faradaic efficiency alone is not sufficient to express the degree of selectivity because the current density of individual species is also an important characteristic of an efficient catalytic CO2R reaction. In addition, as mentioned above, a low faraday efficiency in CO/FA is usually accompanied by a high faraday efficiency in H
2 evolution, which is another valuable product of CO2R in the aqueous solution.
Table 1. Summary of CO2R performances included in this study.
| Base of Catalyst |
FE (%) in FA |
CO2R Condition |
Reference |
| Cu modified with polymeric |
38–45 |
−0.7 VRHE in 0.05 M K2CO3 and 4 mM KCl with 5 sccm CO2 |
[35] |
| boron-doped diamond |
~70 |
−2.1 V (vs. Ag/AgCl) in KCl aqueous solution |
[37] |
| polycrystalline Sn |
~70 |
−1.0 VRHE in 0.1 M KHCO3with 20 sccm CO2 |
[43] |
| sulfur-modified copper |
~80 |
−0.8 VRHE in 0.1 M KHCO3with 20 sccm CO2 |
[44] |
| Cu |
~85 |
−1.6 V vs. ferrocenium voltage in CO2-saturated [EMIM](BF4)/H2O (92/8 v/v) ionic liquid solution |
[45] |
| Cu (1.5 cm × |
| 3 cm) |
~20 |
−0.8 VRHE in 0.1 M KHCO3 with 20 sccm CO2 |
[47] |
| Cu |
~20 |
−1.4 VRHE in 0.5 M KCl with 70 sccm CO2 |
[48] |
| Cu2O@Cu |
~40 |
−0.7 VRHE in 0.1 M KHCO3 with 5 sccm CO2 |
[51] |
| Iron-graphite electrode pair |
~18 |
−0.6 VAg/AgCl with CO2 saturated 0.5 MNaHCO3 in anaerobic sludge digestion process |
[54] |
| Co incovalentorganic frameworks |
1.02 mmol h−1g−1 |
| in CO |
MeCN with triethanolamineas sacrificial reducing agent and Ru(bpy)3Cl2·asphotosensitizerunder simulated sunlight |
[55] |
| In0.6Bi0.2Sn0.2 alloy on a halide perovskite |
~95 |
−1.3 VRHE in 0.5 M KHCO3with 20 sccmCO2under simulated sunlight |
[59] |
| Cu0.5Zn0.5 |
~60 |
−1.3 VRHE in CO2 saturated 0.1 M KHCO3 under simulated sunlight |
[60] |
| SnOx/Sn |
~80 |
−1.7 VAg/AgCl in 0.1 M CO2-saturated KHCO3 |
[61] |
| Pd15Ni85 |
~50 |
−0.5 VRHE in 1.0 M CO2-saturated KHCO3 |
[62] |
| Pb modified with 4-aminomethylbenzene |
~80 |
−1.3 VRHE in 1.0 M CO2-saturated KHCO3 |
[28] |
| N,S-codoped carbon catalysts |
~90 in CO |
−0.6 VRHE in 0.1 M KHCO3with 34 sccm CO2 |
[65] |
| Pd85Cu15/C |
~86 in CO |
−0.9 VRHE in 1.0 M CO2-saturated KHCO3 |
[66] |
defective
β-Bi2O3 double-walled nanotubes |
~90 |
−0.8 VRHE in 0.5M KHCO3with 20 sccm CO2 |
[67] |
| 3.7 nm Pd nanoparticles |
~90 in CO |
−0.9 VRHE in 1.0 M CO2-saturated KHCO3 |
[68] |
| boron-doped Pd catalyst |
~70 |
−0.5 VRHE in 1.0 M CO2-saturated KHCO3 |
[69] |
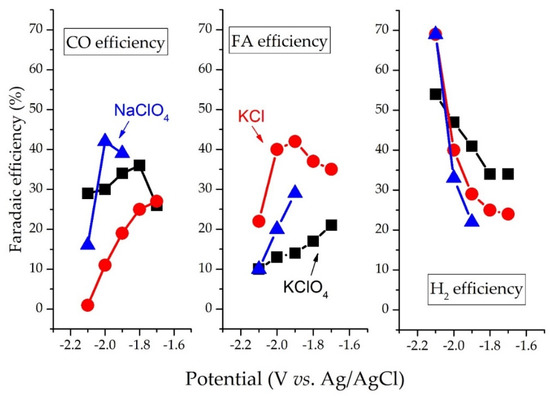
Electrolytes also play a crucial role in determining CO2R selectivity. CO2R over the BDD electrode in the KClO4 electrolyte produces CO, whereas FA is the major product in the KCl electrolyte. This is because ClO4− promotes the adsorption of CO2∙−
intermediates [
37]. Similarly, Eder et al. (2019) have reported that on Ru-based catalysts, hydrosilanes additives and KF stabilized formate intermediates (silylformate) forms form potassium formate with a turnover number of 110 mmol-formate/mmol-Ru [
70]. Note again that the CO2R selectivity is a function of applied voltage, which gradually shifts from
HCOO−/HCOOH to CO/H
2 with increasing overpotentials [
68,
69]. Based on the above consideration, a new approach for effective CO2R by an aluminum hydride-like reductant has been attempted [
71]. The reductant is an organoaluminum complex containing a formal aluminum double bond (dialumene). Weetman et al. (2019) have demonstrated that dialumene improves the selective formation of formic acid equivalent via the dialuminum carbonate complex rather than the conventional aluminum–hydride-based cycle [
71]. Likewise, Zhao et al. (2019) have reported that the
KBH4 reduces
CO2 to
HCO2−
readily, accompanied by the release of activate intermediate species and H
+. Further, CO2R is accelerated by a Cu/Ni bimetal catalyst that effectively regenerated the active boron species [
72].
Briefly, the surface chemistry of non-Cu-family catalysts also strongly influences the selectivity of abiotic CO2R in the aqueous phase. Unlike the Cu-family catalysts, the unique characteristics of non-Cu-family catalysts is that they can be tailored with a specific porous framework. In this configuration, the kinetics in the confined space govern the CO2R selectivity. The localized coordination environment is another factor regulating the overall CO2R selectivity, which can be modified via doping or introducing defects. Along with the modification is the adjustment in the energy levels that further affects the lifetime (stability) of the COR2 intermediate and consequently the CO2R selectivity.
2.2. The Chemistry of Gaseous Phase Abiotic CO2R
Several valuable chemicals such as CO, methane, methanol, low olefins, and long-chain carbohydrates could be produced from the gaseous phase CO2R reaction. To achieve selective CO2R, catalysts that are effective in activating both H
2 and CO
2 and stabilizing surface intermediates are needed. To this end, most catalysts are comprised of metallic sites, which are active in splitting adsorbed H
2 (from H
2 to H*) and exhibit a high affinity toward CO
2 adsorption. Additional modification with alkali or noble metals can further change the surface acidity or aid in the formation of an extra alloy phase in the catalyst [
73]. The metal/support interfacial sites are highly active in CO
2 hydrogenation due to electron perturbation of the metal and partial reduction of metal oxide via the H-spillover mechanism [
74,
75].
Wet impregnation and co-precipitation are the two most frequently used methods to synthesize heterogeneous catalysts for CO
2 hydrogenation [
76,
77]. Nanosized catalysts can be synthesized by flame-spray pyrolysis [
78]. The catalysts made by pyrolyzing metal organic precursors can achieve a complex nanostructure with high surface metal loadings [
79,
80] and the carbon sites can promote the adsorption and activation of CO
2 [
81]. Fixed bed reactor configuration is applied in the evaluation of catalytic performance, in which specific amounts of catalysts are distributed over inert particles, such as SiC or silica, to the minimize hot spot phenomenon. After pre-reduction of the catalysts in H
2 flow, the stream that contains certain ratios of H
2/CO
2/inert gas with a specific gas hourly space velocity (GHSV) is injected into the fixed bed reactor. The CO
2 conversion (X
CO2) and selectivity (S) toward CO and low hydrocarbons are measured from on-line gas chromatography, while the long chain hydrocarbons are collected in a cold trap for further quantification.
2.2.1. Hydrogenation of CO2 to CO and CH4
Under ambient pressure, hydrogenation of CO
2 over most metal catalysts produces either CO or CH
4. The product CO can serve as a feedstock in the methanol synthesis process and the Fischer–Tropsch process for further carbohydrate synthesis [
73]. Conversion of CO
2 to methane could buffer the fluctuations in energy supply via the power-to-gas process that converts excess electricity to H
2 as the reducing agent in CO
2 methanation [
78]. Equations (4) and (5) present the Sabatier reaction and reverse water–gas shift (RWGS), corresponding to the hydrogenation of CO
2 to methane and CO, respectively. Thermodynamically, the former reaction dominates at T < 500 °C, while the latter at T > 500 °C. However, as presented in
Table 2, the selectivity toward CO and methane is greatly altered on the heterogeneous catalysts with the combination of various metals and supports.
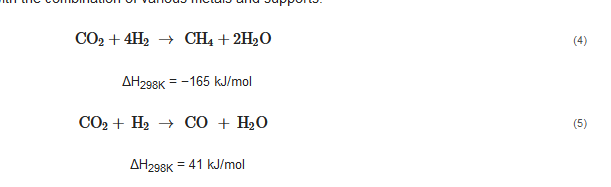
Table 2. Performance of the selected catalysts for CO2 hydrogenation to CO and CH4.
| Catalyst |
T (°C) |
P (MPa) |
H2/CO2/Inert |
GHSV
(mL g−1 h−1) |
XCO2 (%) |
SCO (%) |
SCH4 (%) |
Reference |
| Ru/MnOx |
300 |
0.1 |
22/5/73 |
150,000 |
25 |
10 |
90 |
[78] |
| Ru/Al2O3 |
32 |
6 |
94 |
| Ru/CeO2 |
83 |
1 |
99 |
| Ru/ZnO |
1 |
94 |
6 |
| PtCo/TiO2 |
300 |
0.1 |
67/33/0 |
36,000 |
8.2 |
99 |
1 |
[82] |
| PtCo/CeO2 |
9.1 |
92 |
8 |
| PtCo/ZrO2 |
7.8 |
90 |
11 |
| Co/ZrO2 |
400 |
3.0 |
80/20/0 |
3600 |
92.5 |
< 1 |
> 99 |
[81] |
| Co/SiO2 |
80.1 |
2 |
98 |
| Co/Al2O3 |
77.8 |
3 |
97 |
| Co/TiO2 |
30.9 |
96 |
4 |
| Ni/ZIF-8 a |
420 |
0.1 |
80/20/0 |
15,000 |
43.8 |
97 |
3 |
[81] |
| Fe/ZIF-8 a |
43.8 |
97 |
3 |
| Ni/Fe/ZrO2 |
230 |
0.5 |
80/20/0 |
5000 |
82 |
14 |
86 |
[83] |
| γ-Fe2O3 |
400 |
0.1 |
20/0.1/80 |
1,500,000 |
45 |
30 |
70 |
[84] |
| Ni/MCM b |
400 |
0.1 |
80/20/0 |
90,000 |
73.2 |
8 |
92 |
[85] |
In the presence of noble metals, CO
2 hydrogenation to CO can be realized at a low temperature. This phenomenon is attributed to the lower activation energy of the hydrogenation process on the active sites of metals [
86]. For instance, Dietz et al. (2015) have simulated RWGS at the (111) plane of several metals and found that Ni, Cu, and Rh favor the dissociation of CO
2 → CO + O, while Ag, Pd, and Pt prefer the hydrogenation pathway: CO
2 + H → COOH [
86]. When Pt is loaded on silica and titania, the support itself enhances CO
2 adsorption. The energy change of CO to HCO governs the selectivity toward CO, while the competition for *H
2COH between hydrogenation and C–O bond cleavage affects the preferential production of CH
4 or CH
3OH [
87]. The performance of Pt on RWGS is enhanced by the addition of a potassium promoter, which enables the formation of Pt-O(OH)-K interfacial intermediate that promotes the adsorption of the bicarbonate species, the precursor of CO via the formate pathway. CO
2 conversion in the K-promoted Pt/zeolite system at 500 °C is 27.4%, which is 2-fold greater than the system without K-promoters [
88]. Kattel et al. (2016) further reported that the interfacial sites between PtCo alloy and other reducible oxides (CeO
2, TiO
2 and ZrO
2) are important to stabilizing surface intermediates [
82]. Wang et al. (2015) have studied the mechanisms of CO
2 hydrogenation over Pd/Al
2O
3 and concluded that RWGS and the Sabatier reaction do not take place at the same surface sites [
89].
Ru-based catalysts show great catalytic methanation at a low temperature. Dreyer et al. (2017) have investigated the hydrogenation of CO
2 using Ru-based catalysts dispersed on different metal oxide supports, including Al
2O
3, ZnO, MnO
x, and CeO
2 [
78]. They have found that methanation occurs by partial reduction at metal oxides supports, which increases the coverage of H* but strengthens the C–O bond of CO*. The highest CO
2 conversion (83%) and methane selectivity (99%) at 300 °C is obtained by the Ru/CeO
2 system [
78]. Guo et al. (2018) have demonstrated the metal-support interactions and the effect of H-spillover on CO
2 methanation [
90]. By varying the degree of Ru dispersion from the size of a single atom to nanoparticle (4 nm) on Ru/CeO
2, metal-support interaction is the strongest for a single-atom Ru/CeO
2 that facilitates CO* activation, whereas H-spillover prevails in large Ru clusters and prevents the catalyst from poisoning by enhanced H
2O removal [
90]. Thus, controlling the size of Ru at around 1.2 nm achieves an appropriate balance between the two phenomena, leading to a turnover rate of 1.6-fold and 14-fold greater than single-atom and 4-nm Ru, respectively [
90]. The interactions between Ru and TiO
2 are fortified by syngas pretreatment at 600 °C, which leads to an increase in interfacial sites on Ru-TiO
2 by encapsulation of Ru [
91]. Hydrogenation of CO
2 on nickel-based catalysts has been explored extensively. Dispersing Ni on SiO
2 support is deterministic of selectivity, in which the 10 wt% Ni/SiO
2 is effective in stabilizing the monodentate configured HCOO intermediate, which does not occur on catalysts of low Ni loadings (0.5 wt%) [
92]. Bi et al. (2019) have demonstrated that impregnating Ni on an MCM zeolite with a sodium-free alkaline agent enhances the synergism between Ni and NiO during CO
2 hydrogenation via H
2 adsorption and CO
2 activation, respectively [
85]. The catalyst exhibits remarkable CO
2 conversion (68.3%) and methane selectivity (91.4%) with high stability [
85]. Doping Ni/ZrO
2 with iron enhances the reducibility of Ni and ZrO
2 owning to the electron-donating property of Fe(II), which in turn promotes the synergism effects of Ni-NiO and metal-support interactions [
83,
93].
Based on the above discussions, it is noted that in addition to the reactivity of CO2R catalysts, the chemical environment of the support also plays an important role in CO2R selectivity in the gaseous phase. This is because the gaseous abiotic OC2R is conducted in a high temperature (in comparison with the condition of the aqueous CO2R). In this configuration, the dispersion of CO2R catalysts and consequently their contact with support is highly sensitive to the stability of support. A relative alkaline support such as alumina is beneficial for CO2R efficiency as CO2
is regarded as a weak acid in this configuration. It is thus suggested that in addition to the characteristics of the CO2R catalyst, its dispersion over the support is another issue of concern for rationally engineering a CO2R catalyst with high selectivity and efficiency.
2.2.2. Hydrogenation of CO2 to Methanol
Methanol is a valuable CO
2 reduction product as it can be served as biofuel, building blocks in organic synthesis, and fuels for a methanol-based fuel cell.
Table 3 shows CO
2 conversion to methanol at a pressure of 1–4 MPa over different metallic meal oxide catalysts. Catalyst Cu/Zn/Al
2O
3 plays an important role in the commercial methanol production from syngas. DFT studies reveal that the stabilization of the transition surface species is the key to achieving high selectivity in the syngas conversion. The formate pathway predominates the total process on the surface of Cu/ZnAl
2O
4 and Cu/Zn/Al
2O
3, in which the rate-limiting step is the formation of H
2COO* and H
2COOH* [
94,
95]. The CO production via RWGS is the major byproduct in methanol synthesis. Based on the H/D isotope substitution technique, it is known that methanol synthesis and RWGS occurrence take place at difference surface sites on Cu/ZnO/Al
2O
3, Cu/MgO, Cu/SiO
2, and Pd/SiO
2 [
96]. Karelovic and Ruiz (2015) have suggested that ZnO loads with larger Cu particles tend to suppress the activity of RWGS as the specific methanol formation rate per surface Cu is independent of Cu particle size, while that of CO is enhanced by smaller Cu particles [
97]. Ro et al. (2016) have studied the synergistic effect by dispersing Cu on ZrO
2 and reported that the rate constant of CO
2 conversion on Cu-ZrO
2 interfacial is eight times greater than that on plain Cu [
98]. Phongamwong et al. (2017) have added colloidal silica on Cu/Zn/ZrO
2 as geometric spacers to enhance the stability and performance of the Cu-based catalysts in CO
2 hydrogenation [
99]. The authors have reported that 1% of SiO
2 loading increases the methanol synthesis activity by 26% and retains 12% more activity after emerging from the steam for 96 h at 280 °C.
Table 3. Performance of the selected catalysts for CO2 hydrogenation to methanol.
| Catalyst |
T (°C) |
P (MPa) |
CO2/H2/Inert |
GHSV
(mL g−1 h−1) |
XCO2 (%) |
SCH3OH (%) |
Reference |
| Cu/SiO2 |
250 |
4.1 |
72/24/4 |
3600 |
2.8 |
15 |
[100] |
| Cu/ZnO |
180 |
0.7 |
90/10/0 |
4000 |
0.9 |
94 |
[97] |
| Cu/ZnO/ZrO2/SiO2 |
240 |
2.0 |
30/90/10 |
39,000 |
5.2 |
38 |
[99] |
| Cu/ZnO/ZrO2/MgO/Al2O3 |
250 |
2.0 |
75/25/0 |
2000 |
12.1 |
36 |
[93] |
| Pd/ZnO |
250 |
2.0 |
25/75/0 |
3600 |
10.7 |
60 |
[101] |
| Pd/SiO2 |
250 |
4.1 |
72/24/4 |
3600 |
3.0 |
23 |
[100] |
| Pd-Cu/SiO2 |
6.6 |
34 |
| Pd-Cu/SiO2 |
250 |
5.0 |
75/25/0 |
30,000 |
1.6 |
27 |
[102] |
| MnOx-Co3O4 |
250 |
1.0 |
60/20/20 |
120,000 |
45.1 |
22 |
[103] |
| In2O3 |
270 |
4.0 |
60/20/20 |
15,000 |
1.1 |
55 |
[76] |
| In2O3 |
330 |
4.0 |
60/20/20 |
15,000 |
7.1 |
40 |
| In2O3/ZrO2 |
300 |
5.0 |
80/20/0 |
16,000 |
5.2 |
>99 |
[104] |
Bimetallic Pd, such as PdZn and PdCu, exhibits a high methanol yield similar to Cu-based catalysts even at low temperatures [
73]. The Pd dispersed in ZnO shows exceptional stability attributed to the particle size being maintained at 5 nm even after pre-reduction at 400 °C. While the colloidal dispersion technique is effective in stabilizing the interfacial sites of PdZn, its methanol formation yield is 40-fold greater than the same catalysts synthesized by the traditional wet impregnation method [
101]. PdCu and PdCu
3 bimetallic catalysts exhibit a methanol formation rate of 0.31 μmol gcat
−1 s
−1, which is 3.4-fold and 6.2-fold greater than monometallic Pd and Cu, respectively [
100]. Results of DFT simulation further indicates that the (111) plane of PdCu is highly active in methanol evolution, particularly at the low-coordinated Pd on the stepped surface [
102]. Furthermore, methanol evolution through the formate pathway again is the major catalytic reaction, which can be further promoted by adsorbing small amounts of water to lower the energy barrier through the H-shuttled mechanism [
102]. A novel metal-free In
2O
3 catalyst is also known to exhibit high selectivity in reducing CO
2 to methanol [
76]. The oxygen vacancies on In
2O
3 are important to CO
2 hydrogenation. Specifically, the In
2O
3 with optimal oxygen vacancy exhibits a methanol yield of 3.7 mol kg-cat
−1 h
−1, CO
2 conversion of 7.1%, and methanol selectivity of 40%, respectively, at 330 °C and 4 MPa [
75]. Martin et al. (2016) have reported that the selectivity toward methanol over In
2O
3 approaches 100% in the temperature range of 200–300 °C [
104]. The methanol yield is further promoted by increasing the density of surface oxygen vacancy through Ar sputtering, syngas pretreatment, and ZrO
2 support [
104]. Likewise, incorporating a small fraction of Mn into the spinel Co
3O
4 structure greatly enhances the methanol selectivity in CO
2 hydrogenation, likely attributed to the increase in surface basicity [
103]. In brief, the significance of the thermal stability of the support in the CO2R to methanol is relatively less profound than that in the CO2R to CO and methane. This is because the former is usually carried out in a relatively lower temperature than that of the latter. In this case, the surface basicity becomes much more significant in affecting the reactivity of CO2R catalysts.
2.2.3. Hydrogenation of CO2 to Low and Long Chain Chemicals
CO
2 conversion to lower olefins (C
2–C
4), building blocks, and other long chain hydrocarbons in the gasoline range (C
5–C
11) or diesel range (C
12–C
21) has been explored extensively. Again, catalysts that are reactive in both CO
2 hydrogenation and the Fisher–Tropsch reaction will be capable of achieving the CO
2 conversion objectives [
105].
Table 4 lists the performance of CO
2 hydrogenation to low and long chain hydrocarbons on Fe-based catalysts. Upon CO
2 hydrogenation to different hydrocarbons, the Fe-based catalysts undergo consecutive phase transitions together with the creation of multivalent charges and this surface reconstruction process further diversifies distinct active sites on the catalyst [
84]. The creation of multivalent charges results in the spinel iron (Fe
3O
4) being composed of Fe
3O
4, iron carbides, and α-iron, which enhance the activity of RWGS, carbon chain growth, and olefins’ secondary reactions [
106]. It is therefore suggested that an appropriate fraction of Fe
3O
4 and Fe
5C
2 is necessary for the production of high olefin and paraffin [
107,
108]. Pretreatment of Co-Fe bimetallic under various reducing gases (i.e, H
2, syngas, and CO) is another effective ex-situ modification strategy to precisely control the phase transition [
77]. Under the selected reducing environment, CO activation leads to the formation of CoFe alloy and carburized phases (Co
2C and FeC
x) that shift the selectivity toward low hydrocarbons and oxygenate [
77]. While most Fe-based catalysts produce lower hydrocarbons, delafossite (CuFeO
2) exhibits high selectivity toward long chain hydrocarbons (C
5+) and 85 wt% of the produced long chain hydrocarbons are in the gasoline and diesel range [
109]. Doping alkali metals on Fe-based catalysts is also beneficial to chain growth propagation due to stronger CO
x adsorption [
106]. Compared to Na-free Fe
3O
4, incorporating 1.18 wt% of Na greatly enhances the CO
2 conversion and selectivity toward light olefin from 29.3 to 40.5% and 0.1 to 40.3%, respectively [
110]. The presence of potassium promoter on Fe-Co/Al
2O
3 diminishes the density of hydrogen on metal surfaces, which in turn suppresses the hydrogenation of olefins [
111].
Table 4. Performance of the selected catalysts for CO2 hydrogenation to hydrocarbons.
| Catalyst |
T (°C) |
P (MPa) |
H2/CO2/Inert |
GHSV
(mL g−1 h−1) |
XCO2 (%) |
SCO (%) |
Hydrocarbon Distribution (%) |
Reference |
| CH4 |
C2=-C4= |
C2−-C4− |
C5+ |
| Fe3O4 |
320 |
3 |
72/24/4 |
2000 |
29 |
17 |
60 |
<1 |
36 |
3 |
[110] |
| Na-Fe3O4 |
320 |
3 |
72/24/4 |
2000 |
41 |
14 |
16 |
47 |
8 |
30 |
| Fe2O3 |
350 |
1.5 |
70/23/7 |
1150 |
23 |
21 |
18 |
82 |
[107] |
| K-Fe3O4/Fe2O3 |
270 |
5 |
73/25/2 |
2700 |
37 |
14 |
24 |
42 |
9 |
29 |
[106] |
| CuFeO2 |
300 |
1 |
75/25/0 |
1800 |
18 |
32 |
4 |
31 |
5 |
60 |
[109] |
| CuFe2O4 |
300 |
1 |
75/25/0 |
1800 |
16 |
28 |
38 |
1 |
49 |
11 |
| K-Fe-Co/Al2O3 |
300 |
1.1 |
72/24/4 |
700 |
31 |
18 |
16 |
27 |
6 |
51 |
[111] |
| Co/Fe oxide a |
270 |
0.9 |
72/28/0 |
2000 |
27 |
14 |
82 |
15 |
<1 |
[77] |
| Na-Co/Fe oxide a |
270 |
0.9 |
72/28/0 |
2000 |
23 |
42 |
60 |
29 |
2 |
Pyrolyzed
Fe-MIL-88B b |
400 |
3 |
75/25/0 |
3600 |
46 |
18 |
32 |
23 |
18 |
27 |
[108] |
| K-pyrolyzed Fe-MIL-88B b |
400 |
3 |
75/25/0 |
3600 |
43 |
26 |
32 |
33 |
6 |
19 |
| In2O3/HZSM |
340 |
3 |
73/24/3 |
9000 |
13 |
45 |
1 |
20 |
79 |
[112] |
| Ga2O3/HZSM |
340 |
3 |
73/24/3 |
9000 |
9 |
86 |
5 |
35 |
61 |
| Fe2O3/HZSM |
340 |
3 |
73/24/3 |
9000 |
7 |
74 |
2 |
28 |
71 |
| Na-Fe3O4/HZSM |
350 |
3 |
72/24/4 |
4000 |
33 |
26 |
8 |
18 |
74 |
[105] |
| In2O3-ZrO2/SAPO |
400 |
3 |
73/24/3 |
9000 |
36 |
85 |
4 |
76 |
17 |
3 |
[75] |
Bifunctional catalysts have emerged actively in synthesizing hydrocarbons from CO
2 with great flexibility. Combined In
2O
3 and zeolite (HZSM-5) effectively synthesizes liquid fuels (C
5+) from CO
2 by suppressing undesired C
1 products [
112]. In the process, CO
2 is first hydrogenated to methanol at the reduced site on In
2O
3. Methanol that diffuses to the acidic site of zeolite transforms to hydrocarbons by the hydrocarbon-pool mechanism, which results in 78.6% of C
5+ hydrocarbons [
112]. Replacing the zeolite HZSM-5 by SAPO-34 increases the selectivity toward lower olefins, probably as a result of changes in topology [
74]. Notably, the bifunctional catalyst must be packed in granular instead of powder form as the surface acidity of the zeolite support is another important factor controlling the selectivity of CO
2 hydrogenation toward hydrocarbons [
112]. HZSM-5/Na-Fe
3O
4 composite also effectively converts CO
2 to hydrocarbons in the gasoline range [
110]. The above catalyst composites provide three distinct active sites: Fe
3O
4 sites for RWGS, Fe
5C
2 sites for FTS, and zeolites for oligomerization [
105]. Na-Fe
3O
4/HZSM shows little deactivation; the CO
2 conversion and C
5+ selectivity remain at 27% and 54%, respectively, over 1000 h of operation [
105]. Based on the above discussions, it is noted that the surface chemistry of the support is of equal importance to catalysts in CO2R. Interestingly, surface acidity instead of surface basicity is much more important in this case as the development of long chain chemicals strongly relies on the hydride transfer.
In short, there are advances in the development of heterogeneous catalysts for CO2 hydrogenation in recent years due to a better understanding of the elementary reactions via DFT simulation, catalytic performance of metal-support, and metal-dopant interfacial sites. With appropriate modifications, CO2 can be hydrogenated to CO, CH4, CH3OH, and other low and long chain hydrocarbons. Further challenges are reducing cost and increasing durability of catalysts, reducing cost of renewable hydrogen sources, and carbon dioxide capture.
 +1 credit
+1 credit