 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | KYU SHIK MUN | + 1518 word(s) | 1518 | 2021-07-02 09:03:49 |
Video Upload Options
Cystic fibrosis human organs-on-a-chip can be used in patients with cystic fibrosis (CF). CF is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane regulator (CFTR) gene: the gene product responsible for transporting chloride and bicarbonate ions through the apical membrane of most epithelial cells.
1. Introduction
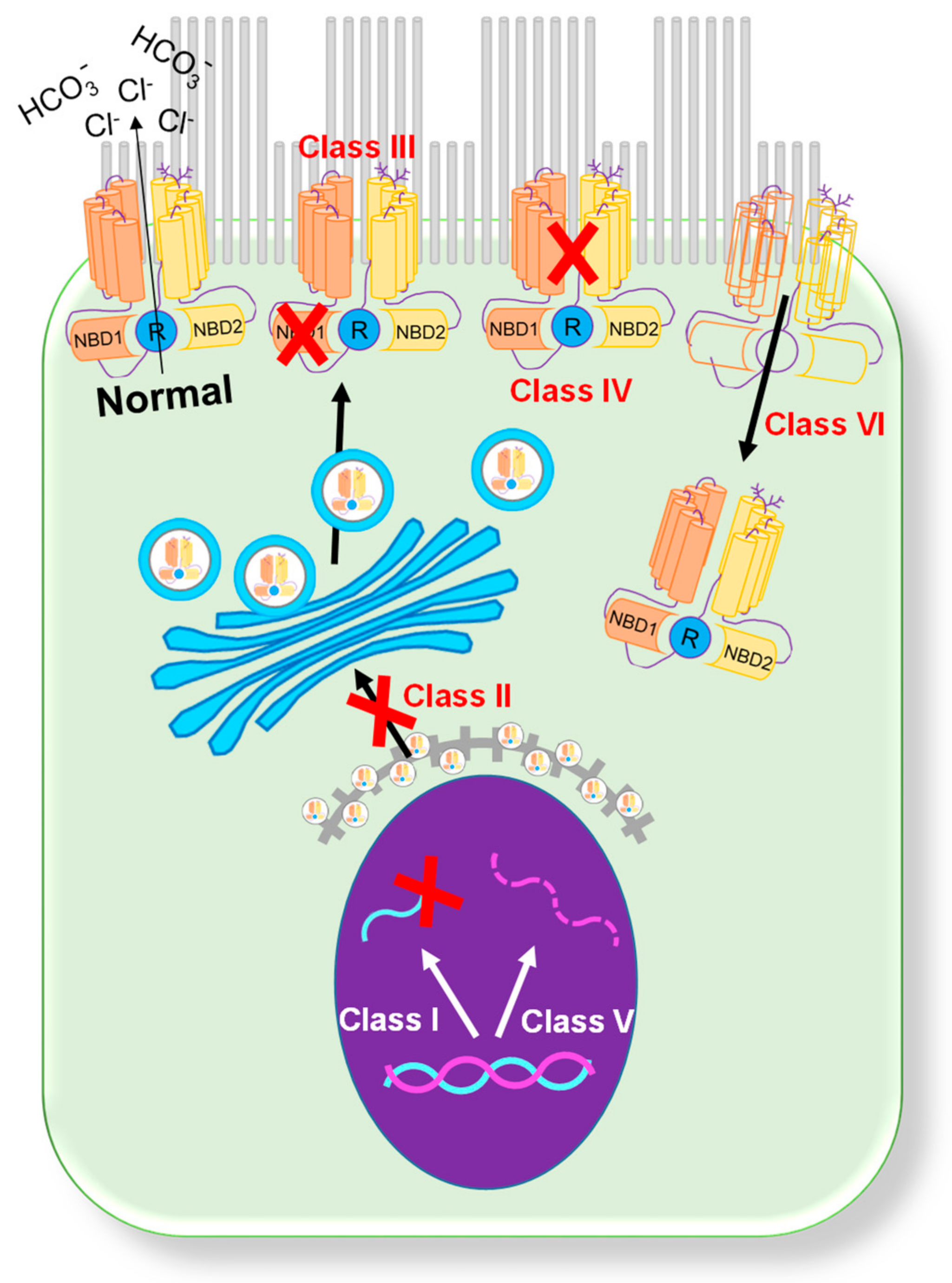 Figure 1. Classification of CFTR mutations[4][11]. A schematic showing six categories of CFTR mutations.
Figure 1. Classification of CFTR mutations[4][11]. A schematic showing six categories of CFTR mutations.2. Organs-on-a-Chip
2.1. Microfluidic-Based Organs-on-a-Chip
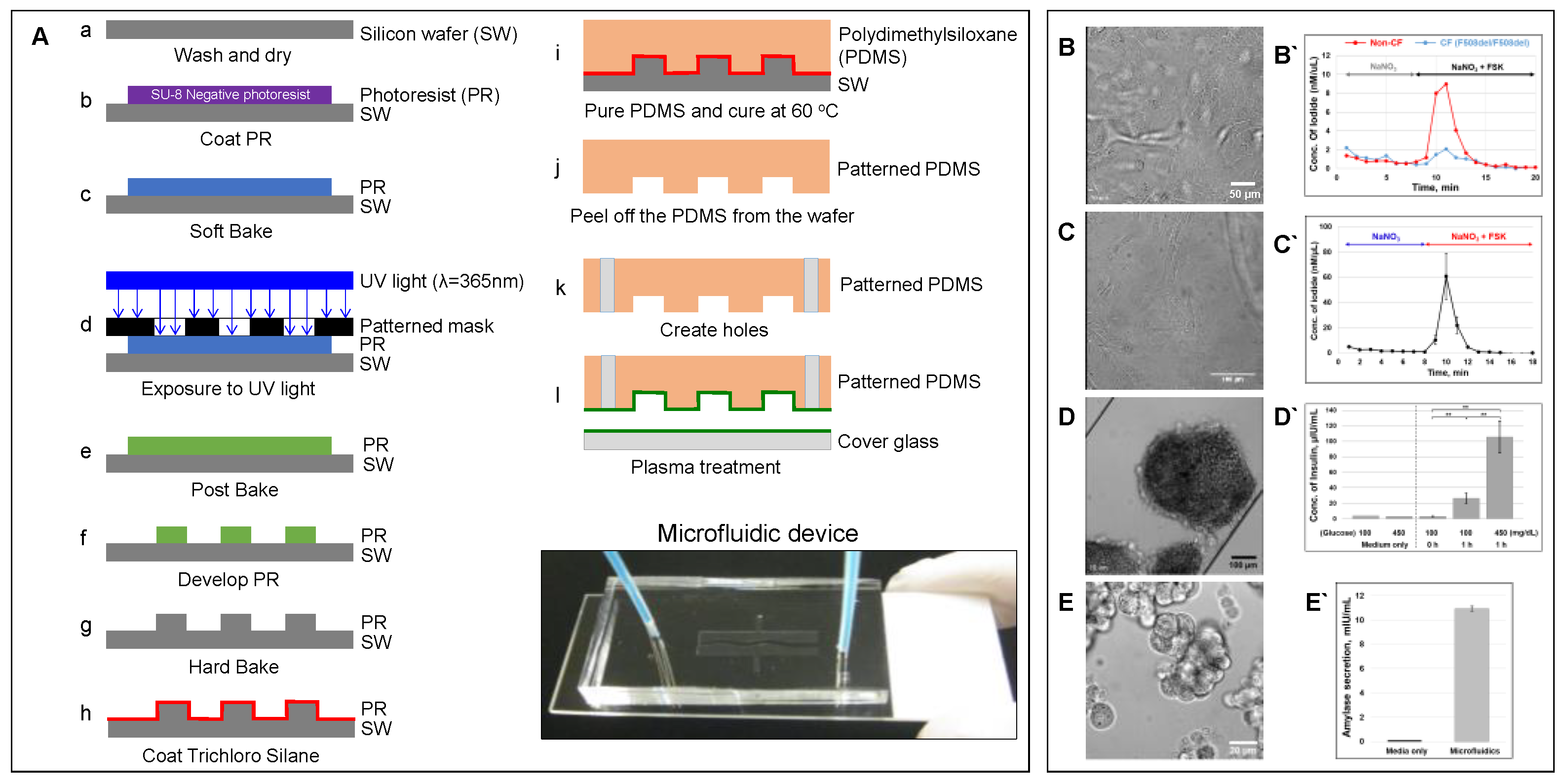 Figure 2. Microfluidic-based organ-on-a-chip[11]. (A) Schematic showing a protocol of standardized photolithography and soft lithography techniques to fabricate a microfluidic device [23]. Patient-derived lung airway epithelial cells obtained from CF (F508del/F508del) and non-CF donors (B), pancreatic ductal epithelial cells (C) [23], aggregated pancreatic islets (D) [23], and aggregated acinar cells (E) were cultured in microfluidic devices and monitored for CFTR function (B`–C` [23]) using an iodide efflux assay. Insulin (D`) [23] and amylase secretion (E`) are also shown. To help cell attachment in the channel, the cells were coated with collagen prior to seeding. (ELISA analysis with p-values: ** < 0.005; number of samples: n = 3; Data represented as mean ± SD).
Figure 2. Microfluidic-based organ-on-a-chip[11]. (A) Schematic showing a protocol of standardized photolithography and soft lithography techniques to fabricate a microfluidic device [23]. Patient-derived lung airway epithelial cells obtained from CF (F508del/F508del) and non-CF donors (B), pancreatic ductal epithelial cells (C) [23], aggregated pancreatic islets (D) [23], and aggregated acinar cells (E) were cultured in microfluidic devices and monitored for CFTR function (B`–C` [23]) using an iodide efflux assay. Insulin (D`) [23] and amylase secretion (E`) are also shown. To help cell attachment in the channel, the cells were coated with collagen prior to seeding. (ELISA analysis with p-values: ** < 0.005; number of samples: n = 3; Data represented as mean ± SD).2.2. Advantages of Organs-on-a-Chip
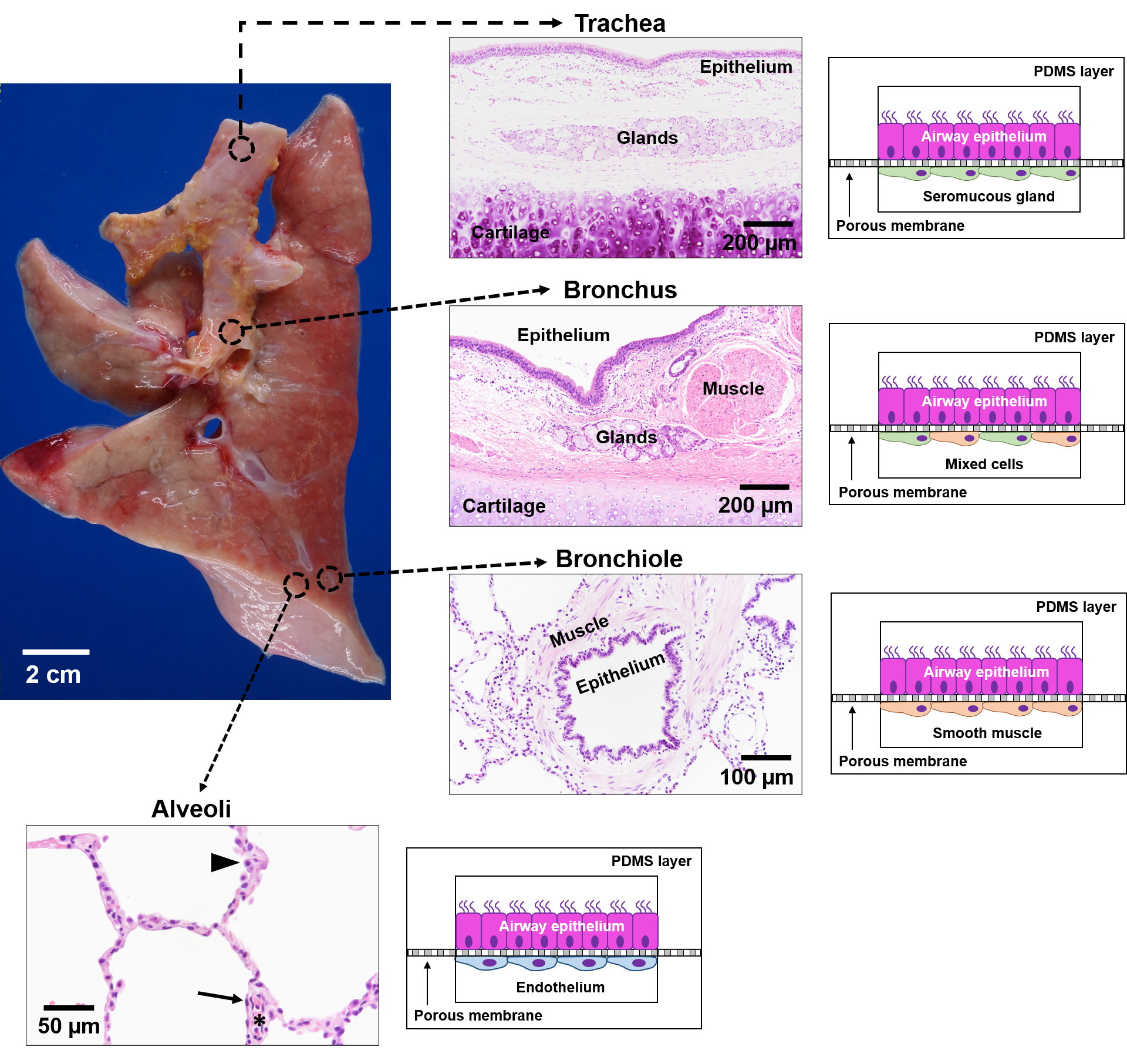
Figure 3. Schematic of lung-on-a-chip models to represent the unique microenvironments throughout the respiratory tract[11]. Four separate microfluidic devices represent four functionally distinct regions of the lung. The alveoli are lined by epithelial type II cells (arrowhead) and type I cells (arrow) that are in close proximity to the endothelial cells lining the capillaries containing red blood cells (*). Each lung-on-a-chip model contains different epithelial cell types specialized for region specific functions with distinct cell populations in the subepithelial compartment to mimic the unique cell-cell interactions seen in spatio-regionally distinct lung microenvironments.
2.3. Limitations of Organs-on-a-Chip
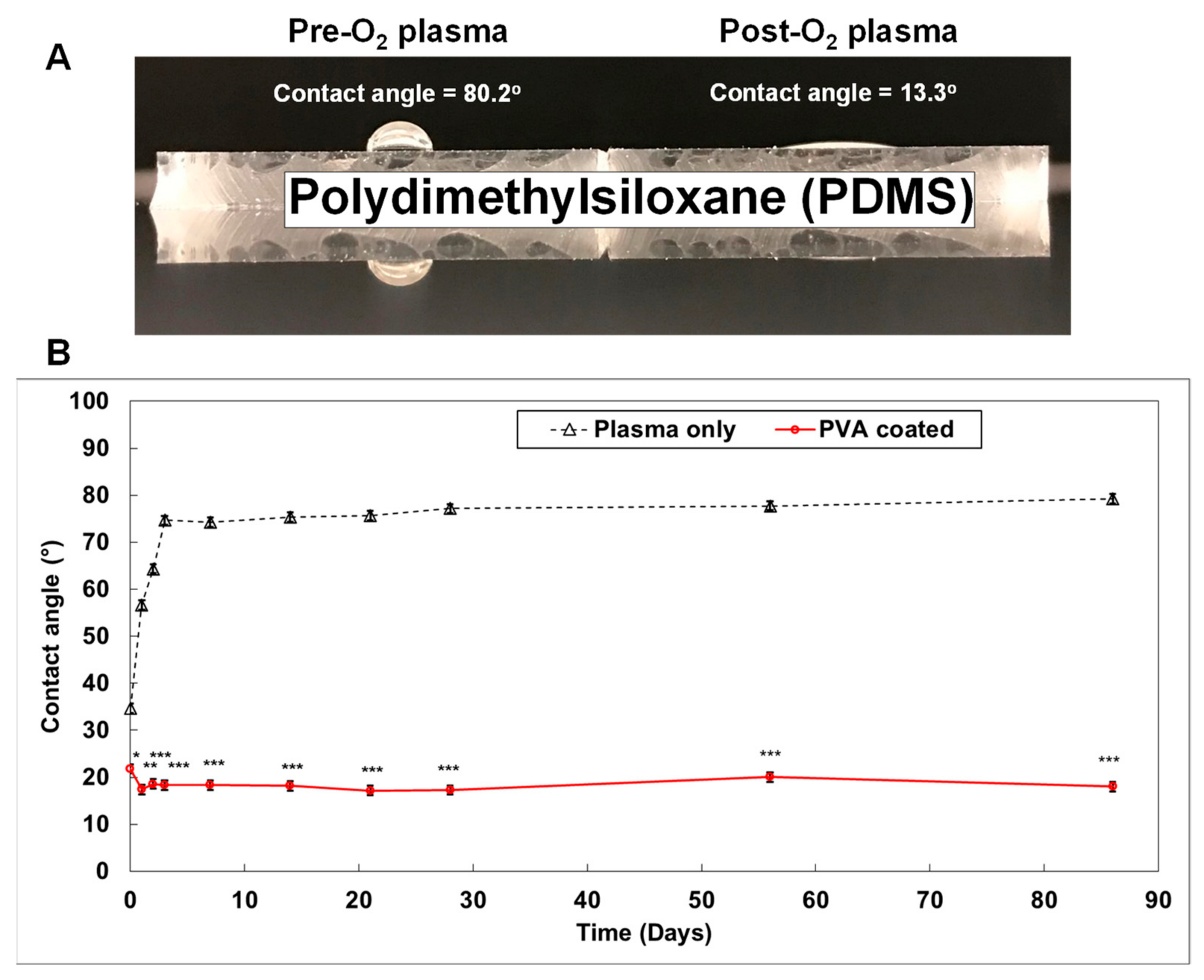 Figure 4. Generating a smart PDMS by modifying a hydrophobic PDMS surface[11]. PDMS is a highly hydrophobic material (A). When the PDMS surface is exposed to oxygen plasma for 30 s, it becomes highly hydrophilic (contact angle = 13.3°). It returns to a hydrophobic status within 3 days (B; black dotted line). However, the polyvinyl alcohol (PVA) coated PDMS surface maintained a hydrophilic status for over 3 months ((B); red solid line). (p-values: * <0.05, ** <0.005, *** <0.0005; number of samples: n = 3 in each condition; data represented as mean ± SD).
Figure 4. Generating a smart PDMS by modifying a hydrophobic PDMS surface[11]. PDMS is a highly hydrophobic material (A). When the PDMS surface is exposed to oxygen plasma for 30 s, it becomes highly hydrophilic (contact angle = 13.3°). It returns to a hydrophobic status within 3 days (B; black dotted line). However, the polyvinyl alcohol (PVA) coated PDMS surface maintained a hydrophilic status for over 3 months ((B); red solid line). (p-values: * <0.05, ** <0.005, *** <0.0005; number of samples: n = 3 in each condition; data represented as mean ± SD).3. Conclusions
References
- Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathologic study. Am. J. Dis. Child. 1938, 56, 344–399.
- Fanen, P.; Wohlhuter-Haddad, A.; Hinzpeter, A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int. J. Biochem. Cell Biol. 2014, 52, 94–102.
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433.
- Harutyunyan, M.; Huang, Y.J.; Mun, K.S.; Yang, F.M.Y.; Arora, K.; Naren, A.P. Personalized medicine in CF: From modulator development to therapy for cystic fibrosis patients with rare CFTR mutations. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, 1529–1543.
- Ratjen, F.; Doring, G. Cystic fibrosis. Lancet 2003, 361, 681–689.
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254.
- Cebotaru, L.; Rapino, D.; Cebotaru, V.; Guggino, W.B. Correcting the cystic fibrosis disease mutant, A455E CFTR. PLoS ONE 2014, 9, e85183.
- Dugueperoux, I.; De Braekeleer, M. The CFTR 3849+10kbC->T and 2789+5G->A alleles are associated with a mild CF phenotype. Eur. Respir. J. 2005, 25, 468–473.
- Beck, S.; Penque, D.; Garcia, S.; Gomes, A.; Farinha, C.; Mata, L.; Gulbenkian, S.; Gil-Ferreira, K.; Duarte, A.; Pacheco, P.; et al. Cystic fibrosis patients with the 3272-26A-->G mutation have mild disease, leaky alternative mRNA splicing, and CFTR protein at the cell membrane. Hum. Mutat. 1999, 14, 133–144.
- Yeh, J.T.; Yu, Y.C.; Hwang, T.C. Structural mechanisms for defective CFTR gating caused by the Q1412X mutation, a severe Class VI pathogenic mutation in cystic fibrosis. J. Physiol. 2019, 597, 543–560.
- Herbert Ogden; Hoyeol Kim; Kathryn Wikenheiser-Brokamp; Anjaparavanda Naren; Kyu Mun; Cystic Fibrosis Human Organs-on-a-Chip. Micromachines 2021, 12, 747, 10.3390/mi12070747.
- Tata, F.; Stanier, P.; Wicking, C.; Halford, S.; Kruyer, H.; Lench, N.J.; Scambler, P.J.; Hansen, C.; Braman, J.C.; Williamson, R.; et al. Cloning the mouse homolog of the human cystic fibrosis transmembrane conductance regulator gene. Genomics 1991, 10, 301–307.
- Colledge, W.H.; Abella, B.S.; Southern, K.W.; Ratcliff, R.; Jiang, C.; Cheng, S.H.; MacVinish, L.J.; Anderson, J.R.; Cuthbert, A.W.; Evans, M.J. Generation and characterization of a delta F508 cystic fibrosis mouse model. Nat. Genet. 1995, 10, 445–452.
- Zhou, L.; Dey, C.R.; Wert, S.E.; DuVall, M.D.; Frizzell, R.A.; Whitsett, J.A. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science 1994, 266, 1705–1708.
- Sun, X.; Sui, H.; Fisher, J.T.; Yan, Z.; Liu, X.; Cho, H.J.; Joo, N.S.; Zhang, Y.; Zhou, W.; Yi, Y.; et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Investig. 2010, 120, 3149–3160.
- Fong, S.L.; Irimura, T.; Landers, R.A.; Bridges, C.D. The carbohydrate of bovine interstitial retinol-binding protein. Prog. Clin. Biol. Res. 1985, 190, 111–128.
- Ostedgaard, L.S.; Rogers, C.S.; Dong, Q.; Randak, C.O.; Vermeer, D.W.; Rokhlina, T.; Karp, P.H.; Welsh, M.J. Processing and function of CFTR-DeltaF508 are species-dependent. Proc. Natl. Acad. Sci. USA 2007, 104, 15370–15375.
- Trezise, A.E.; Szpirer, C.; Buchwald, M. Localization of the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) in the rat to chromosome 4 and implications for the evolution of mammalian chromosomes. Genomics 1992, 14, 869–874.
- Dupuit, F.; Bout, A.; Hinnrasky, J.; Fuchey, C.; Zahm, J.M.; Imler, J.L.; Pavirani, A.; Valerio, D.; Puchelle, E. Expression and localization of CFTR in the rhesus monkey surface airway epithelium. Gene Ther. 1995, 2, 156–163.
- Harris, A. Towards an ovine model of cystic fibrosis. Hum. Mol. Genet. 1997, 6, 2191–2194.
- Fan, Z.; Perisse, I.V.; Cotton, C.U.; Regouski, M.; Meng, Q.; Domb, C.; Van Wettere, A.J.; Wang, Z.; Harris, A.; White, K.L.; et al. A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. JCI Insight 2018, 3, e123529.
- McCarron, A.; Donnelley, M.; Parsons, D. Airway disease phenotypes in animal models of cystic fibrosis. Respir. Res. 2018, 19, 54.
- Mun, K.S.; Arora, K.; Huang, Y.; Yang, F.; Yarlagadda, S.; Ramananda, Y.; Abu-El-Haija, M.; Palermo, J.J.; Appakalai, B.N.; Nathan, J.D.; et al. Patient-derived pancreas-on-a-chip to model cystic fibrosis-related disorders. Nat. Commun. 2019, 10, 3124.
- Gökaltun, A.; Kang, Y.B.; Yarmush, M.L.; Usta, O.B.; Asatekin, A. Simple Surface Modification of Poly(dimethylsiloxane) via Surface Segregating Smart Polymers for Biomicrofluidics. Sci. Rep. 2019, 9, 7377.
- Firpo, G.; Angeli, E.; Repetto, L.; Valbusa, U. Permeability thickness dependence of polydimethylsiloxane (PDMS) membranes. J. Membr. Sci. 2015, 481, 1–8.
- Huh, D.; Kim, H.J.; Fraser, J.P.; Shea, D.E.; Khan, M.; Bahinski, A.; Hamilton, G.A.; Ingber, D.E. Microfabrication of human organs-on-chips. Nat. Protoc. 2013, 8, 2135–2157.
- Trantidou, T.; Elani, Y.; Parsons, E.; Ces, O. Hydrophilic surface modification of PDMS for droplet microfluidics using a simple, quick, and robust method via PVA deposition. Microsyst. Nanoeng. 2017, 3, 16091.
- Mukhopadhyay, R. When PDMS isn’t the best. What are its weaknesses, and which other polymers can researchers add to their toolboxes? Anal. Chem. 2007, 79, 3248–3253.
- Shirure, V.S.; George, S.C. Design considerations to minimize the impact of drug absorption in polymer-based organ-on-a-chip platforms. Lab Chip 2017, 17, 681–690.
- Akther, F.; Yakob, S.B.; Nguyen, N.T.; Ta, H.T. Surface Modification Techniques for Endothelial Cell Seeding in PDMS Microfluidic Devices. Biosensors 2020, 10, 182.

