 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andreas Engert | + 1808 word(s) | 1808 | 2021-04-25 05:32:56 | | | |
| 2 | Karina Chen | Meta information modification | 1808 | 2021-07-08 03:25:45 | | |
Video Upload Options
Hodgkin lymphoma (HL) is a rare neoplasm of the lymphatic system representing one of the most common cancers in young adults. The disease is characterized by a low number of malignant cells (Hodgkin- and Reed-Sternberg cells) deriving from B-lymphocytes and an extensive inflammatory microenvironment This unique histopathological picture and its pathogenesis are still only partially understood.
1. Epidemiology and Risk Factors
The incidence of Hodgkin lymphoma is estimated to be 2–3 per 100,000 individuals per year with a peak in the third decade of life and a second peak around 60 years.[1]
Various factors seem to favor the occurrence of HL. The significantly increased risk of identical twins strongly indicates the role of genetics in HL. Epstein–Barr virus (EBV) is detected in nearly 45% of HL patients.[2] EBV infection seems to be a relevant factor in pathogenesis and HIV infection associates with elevated risk but both are not sole contributors.[3]
2. Histopathology
Histopathologically, 95% of HL cases are classified as classical (cHL) including the subtypes nodular sclerosing, mixed cellularity, lymphocyte-rich and lymphocyte-depleted HL. In 5% of cases, Nodular lymphocyte-predominant Hodgkin Lymphoma (NLPHL) is diagnosed.[4] While cHL is characterized by the presence of CD30-expressing Hodgkin and Reed-Sternberg (HRS) cells surrounded by a mixture of inflammatory cells, the malignant cells in NPLHL are termed lymphocyte predominant (LP) cells. They are positive for CD20 and lack CD30. These cells are surrounded by mature lymphocytes.
3. Pathophysiology
cHL is a B-cell lymphoma of germinal center origin that has lost its B-cell phenotype. HRS cells harbor clonal rearrangements of hypermutated, class-switched immunoglobulin genes, which result in non-functional immunoglobulin genes lacking the expression of cell surface B-cell receptor. In a healthy B-cell, this should lead to apoptosis, however in HL, these cells appear to be “rescued” from apoptosis by additional oncogenic events.[5][6]
In addition to continued intracellular survival and proliferation signaling, HRS cells require a peculiar cellular microenvironment (TME). A deeper insight into the mechanisms by which HRS cells orchestrate their microenvironment and evade T-cell and natural killer (NK)-cell-mediated antitumoral immune response significantly contributed to the understanding of HL biology providing new treatment approaches.[7]
Genomic analyses of HRS cells have shown that certain genetic aberrations significantly contribute to their altered interaction with the inflammatory microenvironment. Reduced MHC class I or II presentation on their surface, either by downregulation, loss-of-function mutations (B2M) or translocations (CIITA) impair antigen presentation.[8] HRS cells frequently harbor an increased copy number of genes located on chromosome 9p24.1 encoding programmed death receptor ligands PDL1 and 2. Overexpressed PDL1/L2 interacts with PD1 on many types T cells in the HL microenvironment. Furthermore, NK-cell mediated antitumoral immune response might be inhibited by aberrant expression of MICA in HRS cells.[9] The complex interactions of HRS cells with their inflammatory environment are not completely understood.[10]
4. Diagnosis and Staging
Removal and histopathological analysis of a lymph node is the method of choice for diagnosis. Since diagnosis can be complicated, pathological expert review is recommended. Detailed patient history including evaluation of B-Symptoms, clinical examination and imaging procedures including contrast-enhanced CT (ceCT) and 18FDG-Positron emission tomography (PET/CT), if available, are regarded as mandatory for initial staging. Moreover, PET/CT is highly sensitive to detect bone marrow involvement and allows omission of the bone marrow biopsy in the case of PET negativity.[11] Individual stages of HL are differentiated using the modified Ann-Arbor classification and defined risk factors. Risk stratification varies between Europe and North America. In Europe, three different risk groups are relevant including early-stage favorable, early-stage unfavorable and advanced stage HL
Since chemo- and radiotherapy can potentially affect the fertility of patients, all patients should be offered the possibility of fertility maintenance measures if family planning is not finalized. [12]
5. Treatment strategies
First-Line treatment of cHL
The current standard for early-stage favorable cHL consists of two cycles of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) and consolidation involved-site (IS) radiotherapy with 20 Gray (Gy). Long time follow-up analyses of GHSG HD10 trial showed progression free survival rates of 87% and overall survival rates of 94% for this approach.[13] Omitting parts of the ABVD regimen and PET-guided omission of radiotherapy resulted in a significantly reduced tumor control.[14]
Patients with initial diagnosis of early-stage unfavorable HL are usually treated with four cycles of polychemotherapy. Depending on response and intensity of systemic therapy, a consolidation radiotherapy is applied. The recently completed GHSG HD17 trial showed that patients who are PET-negative after 2 cycles of eBEACOPP + 2 cycles of ABVD (“2+2”) do not require consolidating 30 Gy INRT radiotherapy (involved node RT).[15]
Patients with advanced-stage cHL are more likely to relapse or have refractory disease and therefore require a more intensive treatment. Internationally, treatment with six cycles of ABVD and consecutive PET-adopted radiotherapy remains the standard therapy for advanced stages in many countries.[16] However, the eBEACOPP regime results in more frequent and long-lasting remissions. A meta-analysis comprising almost 10,000 patients from 14 different studies comparing ABVD with eBEACOPP showed a significant survival benefit of 7% compared to ABVD in the advanced stages of HL.[17] Therefore, the treatment of advanced stage HL with eBEACOPP represents the standard of care by EORTC, LYSA and GHSG. Based on the results of the GHSG HD18 study achieving a 5-year PFS of 90–92%, the current GHSG standard for patients in advanced stages under 60 years of age consists of four or six cycles of eBEACOPP—depending on the early PET-based response after the second cycle—followed by PET-based radiotherapy. [ref When given after two initial cycles of eBEACOPP, a de-escalation of the treatment by switching to ABVD appears be feasible, as shown by the LYSA AHL2011 trial.[18] The ongoing GHSG 21 trial evaluates a different approach to further reducing chemotherapy- associated acute and long-term toxicity in advanced stage cHL: the combination of BV with a modified eBEACOPP regimen termed BrECADD. The Echelon-1 trial aimed improving tumor control in advanced cHL patients by adding the anti-CD30 immunoconjugate Brentuximab Vedotin to AVD. This resulted in improved tumor control over ABVD, however also showing an significantly increased toxicity.[19]
Figure 1 summarizes the different treatment approaches in advanced-stage cHL.
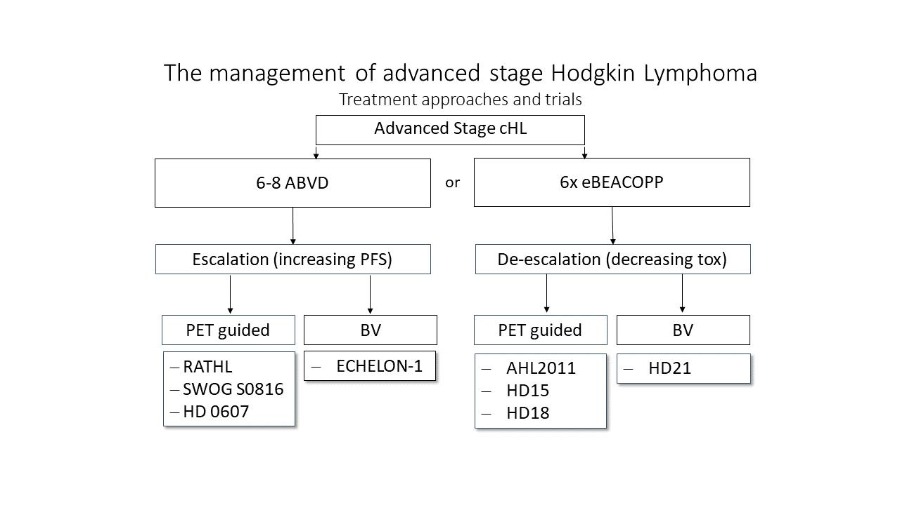
Figure 1: Treatment options overview for advanced cHL.[20] The figure displays treatment approaches reviewed in clinical trials. Either escalating therapy to improve PFS by PET-guided therapies or new drugs (BV) or de-escalation of efficient but toxic treatment approaches guided by PET/CT or including new targeted drugs.
Second Line treatment
Following front-line treatment failure, the application of an intensive salvage chemotherapy followed by high-dose chemotherapy (HDCT) and autologous stem cell transplant (ASCT) is regarded as the standard of care. This approach results in a cure rate of about 50%.[21] Based on the results of the AETHERA trial, consolidating use of brentuximab vedotin after ASCT results in significant improvement of disease control and thus is approved for high-risk patients.[22]
Third Line treatment
With the approval of the anti-CD30 immunoconjugate BV and the anti-PD1 antibodies Nivolumab and Pembrolizumab, new treatment options have become available and resulted in high response rates even in chemotherapy-refractory and heavily pretreated cHL patients.
In the pivotal phase II trial, evaluating BV as monotherapy in cHL patients with relapse after ASCT, a response rate of 75% and a median PFS of 9.3 months in cHL patients were documented.[23] In patients achieving a complete response with BV, 34% of these patients treated in a phase II trial, a 5-year PFS of 52% was documented indicating a potentially curative role of BV in a minority of patients with r/r cHL after ASCT.[24] The idea to re-activate exhausted T-lymphocytes in the microenvironment of cHL by blockade of the checkpoint molecule PD1 revolutionized the treatment of r/r cHL: the application of Nivolumab and Pembrolizumab in the pivotal phase I/II trials resulted in response rates of about 70% and a median PFS of 13-15 months in patients with prior ASCT and Brentuximab.[25][26] Although the rate of complete responses achieved with Nivolumab and Pembrolizumab in these trials was rather low and most patients relapsed within the follow-up period, excellent overall survival rates of over 80% after 2 years could be documented. The question whether BV or an anti-PD-1 antibody should be used in case of relapse after ASCT was addressed in the Keynote-204 trial (NCT02684292): With a median follow-up of 24.7 months a significant PFS improvement in the pembrolizumab vs BV arm was observed so that Pembrolizumab should be discussed as a new standard for patients with relapse after autologous SCT.[27]
Figure 2 shows a therapy algorithm for relapsed/refractory HL.
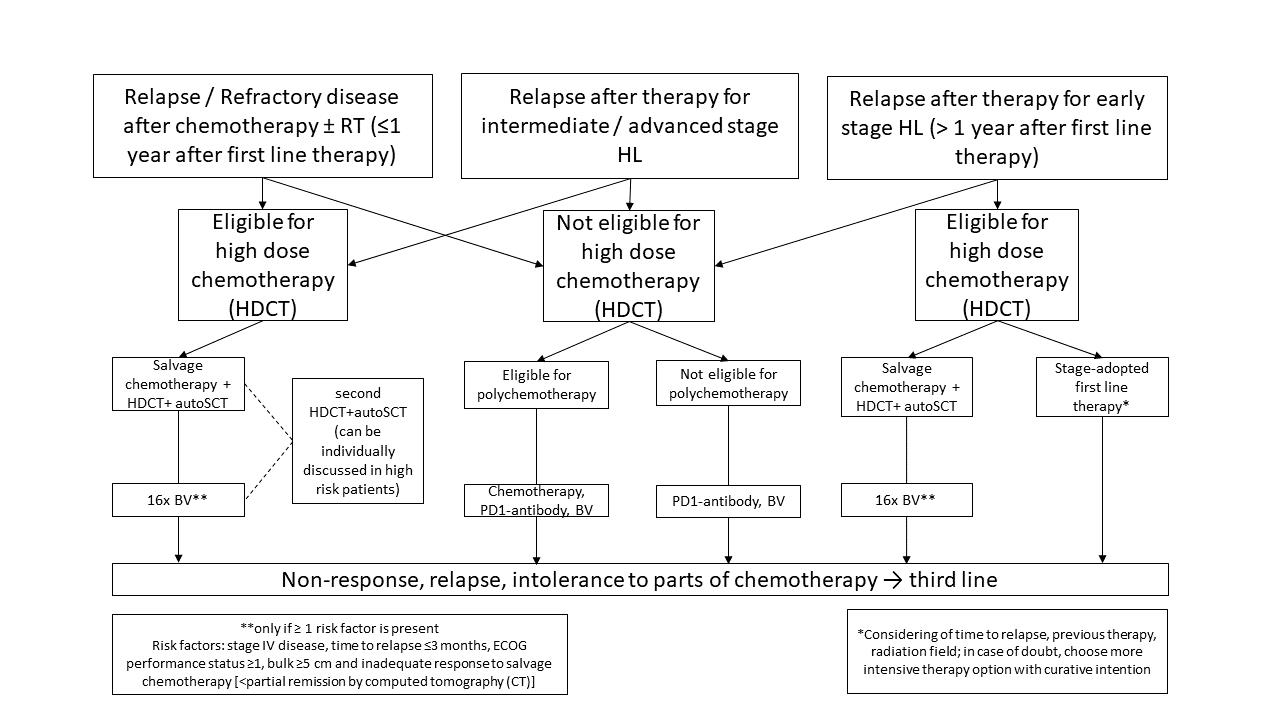
Figure 2: Treatment options overview for r/rcHL. For in depth-disussion please refer to Momotow et al. [20]
Special cohorts: NLPHL
Nodular lymphocyte-predominant HL (NLPHL) accounts for approximately 5% of all HL cases. Pathological and clinical characteristics differ from cHL. Histopathologically, the malignant lymphocyte predominant cells in NLPHL are consistently positive for CD20, but often lack CD30. Clinically, most cases are diagnosed in early stages and the course is usually indolent. However, a tendency towards late relapses and histological transformation into aggressive B-cell non-Hodgkin lymphoma (B-NHL) has been described.[28] At most institutions, the standard treatment for stage IA NLPHL without clinical risk factors consists of limited-field RT alone. Different retrospective studies have demonstrated that the addition of chemotherapy does not further improve the results obtained with RT alone.[29] All other stages are domain of combined modality treatments consisting of chemotherapy and radiotherapy approaches, identical to those used for classical Hodgkin lymphoma in first line treatment. Overall, patients with NLPHL have an excellent outcome after stage-adapted first-line treatment. In relapse settings, antibody treatment against the protein CD20 can be considered.
Special cohorts: Elderly patients
The successful progress in treatment results in patients below 60 years of age could not be translated to the elderly patient cohort. Elderly patients tolerate intensive chemotherapy regimens less well. Additionally, the course of the disease in older patients often appears more aggressive than in the younger patient cohort.[30] Based on the available trial data elderly patients with early-stage favorable cHL and eligible for chemotherapy should be treated with two cycles of ABVD and additional 20 Gy radiotherapy similar to the younger patient cohort.[31] Early-unfavorable cHL in patients older than 60 years should be treated with two cycles of ABVD and two cycles of AVD with consecutive radiotherapy ad 30 Gy IS-RT.[32] Due to toxicity, elderly patients should not be treated with more than two cycles of ABVD including Bleomycin due to the higher risk for long term lung toxicity.[33] Instead, treatment with two cycles ABVD and a further four cycles of AVD and subsequent irradiation of PET-positive residues > 1.5 cm can be regarded as the treatment of choice in elderly patients with advanced-stage cHL.
6. Outlook
The introduction of Brentuximab Vedotin and PD-1 inhibitors such as Pembrolizumab and Nivolumab has opened up new treatment options for Hodgkin lymphoma.[34] The introduction of checkpoint inhibitors and their success in the treatment of relapsed/refractory cHL suggest that an implementation of these drugs in first-line treatment approaches enables new, less toxic and effective treatment strategies in the near future.
References
- Hans H. Storm; Åsa Klint; Laufey Tryggvadottir; Mette Gislum; Gerda Engholm; Freddie Bray; Timo Hakulinen; Trends in the survival of patients diagnosed with malignant neoplasms of lymphoid, haematopoietic, and related tissue in the Nordic countries 1964–2003 followed up to the end of 2006. Acta Oncologica 2010, 49, 694-712, 10.3109/02841861003631495.
- G. Pallesen; S.J. Hamilton-Dutoit; Martin Rowe; L.S. Young; Expression of Epstein-Barr virus latent gene products in tumour cells of Hodgkin's disease. The Lancet 1991, 337, 320-322, 10.1016/0140-6736(91)90943-j.
- Raúl U Hernández-Ramírez; Meredith S Shiels; Robert Dubrow; Eric A Engels; Cancer risk in HIV-infected people in the USA from 1996 to 2012: a population-based, registry-linkage study. The Lancet HIV 2017, 4, e495-e504, 10.1016/s2352-3018(17)30125-x.
- Ralf Küppers; Andreas Engert; Martin-Leo Hansmann; Hodgkin lymphoma. Journal of Clinical Investigation 2012, 122, 3439-3447, 10.1172/jci61245.
- Holger Kanzler; Ralf Küppers; Sabine Helmes; Hans-Heinrich Wacker; Andreas Chott; Martin-Leo Hansmann; Klaus Rajewsky; Hodgkin and Reed-Sternberg–like cells in B-cell chronic lymphocytic leukemia represent the outgrowth of single germinal-center B-cell–derived clones: potential precursors of Hodgkin and Reed-Sternberg cells in Hodgkin's disease. Blood 2000, 95, 1023-1031, 10.1182/blood.v95.3.1023.003k07_1023_1031.
- Ines Schwering; Andreas Bräuninger; Ulf Klein; Berit Jungnickel; Marianne Tinguely; Volker Diehl; Martin-Leo Hansmann; Riccardo Dalla-Favera; Klaus Rajewsky; Ralf Küppers; et al. Loss of the B-lineage–specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003, 101, 1505-1512, 10.1182/blood-2002-03-0839.
- W. Robert Liu; Margaret A. Shipp; Signaling pathways and immune evasion mechanisms in classical Hodgkin lymphoma. Blood 2017, 130, 2265-2270, 10.1182/blood-2017-06-781989.
- Christian Steidl; Sohrab P. Shah; Bruce W. Woolcock; Lixin Rui; Masahiro Kawahara; Pedro Farinha; Nathalie A. Johnson; Yongjun Zhao; Adele Telenius; Susana Ben Neriah; et al.Andrew McPhersonBarbara MeissnerUjunwa Okoye-OkaforArjan DiepstraAnke Van Den BergMark SunGillian LeungSteven JonesJoseph M. ConnorsDavid G. HuntsmanKerry J. SavageLisa M. RimszaDouglas E. HorsmanLouis M. StaudtUlrich SteidlMarco A. MarraRandy D. Gascoyne MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011, 471, 377-381, 10.1038/nature09754.
- Joerg Kessler; Katrin S. Reiners; Maike Sauer; Andreas Engert; Elke Pogge Von Strandmann; NK Cells in Hodgkin Lymphoma Are Impaired but Can Be Activated. Blood 2011, 118, 2182-2182, 10.1182/blood.v118.21.2182.2182.
- Fathima Zumla Cader; Ron C. J. Schackmann; Xihao Hu; Kirsty Wienand; Robert Redd; Bjoern Chapuy; Jing Ouyang; Nicole Paul; Evisa Gjini; Mikel Lipschitz; et al.Philippe ArmandDavid WuJonathan R. FrommDonna NeubergX. Shirley LiuScott J. RodigMargaret A. Shipp Mass cytometry of Hodgkin lymphoma reveals a CD4+ regulatory T-cell–rich and exhausted T-effector microenvironment. Blood 2018, 132, 825-836, 10.1182/blood-2018-04-843714.
- Tarec Christoffer El-Galaly; Francesco D'amore; Karen Juul Mylam; Peter De Nully Brown; Martin Bøgsted; Anne Bukh; Lena Specht; Annika Loft; Victor Iyer; Karin Hjorthaug; et al.Anne Lerberg NielsenIlse ChristiansenCharlotte MadsenHans-Erik JohnsenMartin Hutchings Routine Bone Marrow Biopsy Has Little or No Therapeutic Consequence for Positron Emission Tomography/Computed Tomography–Staged Treatment-Naive Patients With Hodgkin Lymphoma. Journal of Clinical Oncology 2012, 30, 4508-4514, 10.1200/jco.2012.42.4036.
- Karolin Behringer; Horst Mueller; Helen Goergen; Indra Thielen; Angelika Diana Eibl; Volker Stumpf; Carsten Wessels; Martin Wiehlpütz; Johannes Rosenbrock; Teresa Halbsguth; et al.Katrin S. ReinersThomas SchoberJorg H. RennoMichael von WolffKatrin Van Der VenMarietta KuehrMichael FuchsVolker DiehlAndreas EngertPeter Borchmann Gonadal Function and Fertility in Survivors After Hodgkin Lymphoma Treatment Within the German Hodgkin Study Group HD13 to HD15 Trials. Journal of Clinical Oncology 2013, 31, 231-239, 10.1200/jco.2012.44.3721.
- Stephanie Sasse; Paul J. Bröckelmann; Helen Goergen; Annette Plütschow; Horst Müller; Stefanie Kreissl; Carolin Buerkle; Sven Borchmann; Michael Fuchs; Peter Borchmann; et al.Volker DiehlAndreas Engert Long-Term Follow-Up of Contemporary Treatment in Early-Stage Hodgkin Lymphoma: Updated Analyses of the German Hodgkin Study Group HD7, HD8, HD10, and HD11 Trials. Journal of Clinical Oncology 2017, 35, 1999-2007, 10.1200/jco.2016.70.9410.
- Karolin Behringer; Helen Goergen; Felicitas Hitz; Josée M Zijlstra; Richard Greil; Jana Markova; Stephanie Sasse; Michael Fuchs; Max S Topp; Martin Soekler; et al.Stephan MathasJulia MeissnerMartin WilhelmPeter KochHans-Walter LindemannEnrico SchalkRobert SemrauJan KrizTom VielerMartin BentzElisabeth LangeRolf MahlbergAndre HasslerMartin VogelhuberDennis HahnJörg MezgerStefan KrauseNicole SkoetzBoris BöllBastian von TresckowVolker DiehlMichael HallekPeter BorchmannHarald SteinHans EichAndreas Engert Omission of dacarbazine or bleomycin, or both, from the ABVD regimen in treatment of early-stage favourable Hodgkin's lymphoma (GHSG HD13): an open-label, randomised, non-inferiority trial. The Lancet 2015, 385, 1418-1427, 10.1016/s0140-6736(14)61469-0.
- Borchmann P, Plütschow A, Kobe C, Greil R, Meissner J, Topp MS, Ostermann H, Dierlamm J, Mohm J, Thiemer J, Sökler M, Kerkhoff A, Ahlborn M, Halbsguth TV, Martin S, Keller U, Balabanov S, Pabst T, Vogelhuber M, Hüttmann A, Wilhelm M, Zijlstra JM, Moccia A, Kuhnert G, Bröckelmann PJ, von Tresckow B, Fuchs M, Klimm B, Rosenwald A, Eich H, Baues C, Marnitz S, Hallek M, Diehl V, Dietlein M, Engert A; PET-guided omission of radiotherapy in early-stage unfavourable Hodgkin lymphoma (GHSG HD17): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncology 2021, Feb;22(2):, 223-234, 10.1016/S1470-2045(20)30601-X..
- Andrea Gallamini; Andrea Rossi; Caterina Patti; Marco Picardi; Alessandra Romano; Maria Cantonetti; Sara Oppi; Simonetta Viviani; Silvia Bolis; Livio Trentin; et al.Guido GiniRoberta BattistiniStephane ChauvieRoberto SorasioChiara PavoniRoberta ZanottiMichele CimminielloCorrado SchiavottoPiera VieroAntonino MuléFederico FallancaUmberto FicolaCorrado TarellaLuca GuerraAlessandro Rambaldi Consolidation Radiotherapy Could Be Safely Omitted in Advanced Hodgkin Lymphoma With Large Nodal Mass in Complete Metabolic Response After ABVD: Final Analysis of the Randomized GITIL/FIL HD0607 Trial. Journal of Clinical Oncology 2020, 38, 3905-3913, 10.1200/jco.20.00935.
- Nicole Skoetz; Sven Trelle; Michaela Rancea; Heinz Haverkamp; Volker Diehl; Andreas Engert; Peter Borchmann; Effect of initial treatment strategy on survival of patients with advanced-stage Hodgkin's lymphoma: a systematic review and network meta-analysis. The Lancet Oncology 2013, 14, 943-952, 10.1016/s1470-2045(13)70341-3.
- René-Olivier Casasnovas; Reda Bouabdallah; Pauline Brice; Julien Lazarovici; Hervé Ghesquieres; Aspasia Stamatoullas; Jehan Dupuis; Anne-Claire Gac; Thomas Gastinne; Bertrand Joly; et al.Krimo BouabdallahEmmanuelle Nicolas-VirelizierPierre FeugierFranck MorschhauserRichard DelarueHassan FarhatPhilippe QuittetAlina Berriolo-RiedingerAdrian TempesculVéronique EdelineHervé MaisonneuveLuc-Matthieu ForneckerThierry LamyAlain DelmerPeggy DartiguesLaurent MartinMarc AndréNicolas MounierAlexandra Traverse-GlehenMichel Meignan PET-adapted treatment for newly diagnosed advanced Hodgkin lymphoma (AHL2011): a randomised, multicentre, non-inferiority, phase 3 study. The Lancet Oncology 2019, 20, 202-215, 10.1016/s1470-2045(18)30784-8.
- Joseph M. Connors; Wojciech Jurczak; David J. Straus; Stephen M. Ansell; Won S. Kim; Andrea Gallamini; Anas Younes; Sergey Alekseev; Árpád Illés; Marco Picardi; et al.Ewa Lech-MarandaYasuhiro OkiTatyana FeldmanPiotr SmolewskiKerry J. SavageNancy L. BartlettJan WalewskiRobert ChenRadhakrishnan RamchandrenPier Luigi ZinzaniDavid CunninghamAndras RostaNeil C. JosephsonEric SongJessica SachsRachael LiuHina A. JolinDirk HuebnerJohn Radford Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. New England Journal of Medicine 2018, 378, 331-344, 10.1056/nejmoa1708984.
- Jesko Momotow; Sven Borchmann; Dennis Eichenauer; Andreas Engert; Stephanie Sasse; Hodgkin Lymphoma—Review on Pathogenesis, Diagnosis, Current and Future Treatment Approaches for Adult Patients. Journal of Clinical Medicine 2021, 10, 1125, 10.3390/jcm10051125.
- Norbert Schmitz; Beate Pfistner; Michael Sextro; Markus Sieber; Angelo M Carella; Matthias Haenel; Friederike Boissevain; Reinhart Zschaber; Peter Müller; Hartmut Kirchner; et al.Andreas LohriSusanne DeckerBettina KochDirk HasencleverAnthony H GoldstoneVolker Diehl Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin's disease: a randomised trial. The Lancet 2002, 359, 2065-2071, 10.1016/s0140-6736(02)08938-9.
- Craig H Moskowitz; Auayporn Nademanee; Tamas Masszi; Edward Agura; Jerzy Holowiecki; Muneer Abidi; Andy I Chen; Patrick Stiff; Alessandro M Gianni; Angelo Carella; et al.Dzhelil OsmanovVeronika BachanovaJohn SweetenhamAnna SuredaDirk HuebnerEric SieversAndy ChiEmily K LarsenNaomi N HunderJan Walewski Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin's lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet 2015, 385, 1853-1862, 10.1016/s0140-6736(15)60165-9.
- Anas Younes; Ajay Gopal; Scott E. Smith; Stephen M. Ansell; Joseph D. Rosenblatt; Kerry J. Savage; Radhakrishnan Ramchandren; Nancy L. Bartlett; Bruce D. Cheson; Sven De Vos; et al.Andres Forero-TorresCraig H. MoskowitzJoseph M. ConnorsAndreas EngertEmily K. LarsenDana A. KennedyEric SieversRobert Chen Results of a Pivotal Phase II Study of Brentuximab Vedotin for Patients With Relapsed or Refractory Hodgkin's Lymphoma. Journal of Clinical Oncology 2012, 30, 2183-2189, 10.1200/jco.2011.38.0410.
- Robert Chen; Ajay Gopal; Scott E. Smith; Stephen M. Ansell; Joseph D. Rosenblatt; Kerry J. Savage; Joseph M. Connors; Andreas Engert; Emily K. Larsen; Dirk Huebner; et al.Abraham FongAnas Younes Five-year survival and durability results of brentuximab vedotin in patients with relapsed or refractory Hodgkin lymphoma. Blood 2016, 128, 1562-1566, 10.1182/blood-2016-02-699850.
- Philippe Armand; Andreas Engert; Anas Younes; Michelle Fanale; Armando Santoro; Pier Luigi Zinzani; John M. Timmerman; Graham Collins; Radhakrishnan Ramchandren; Jonathon B. Cohen; et al.Jan Paul De BoerJohn KuruvillaKerry J. SavageMarek TrnenyMargaret A. ShippKazunobu KatoAnne SumbulBenedetto FarsaciStephen M. Ansell Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial. Journal of Clinical Oncology 2018, 36, 1428-1439, 10.1200/jco.2017.76.0793.
- Robert Chen; Pier Luigi Zinzani; Michelle A. Fanale; Philippe Armand; Nathalie A. Johnson; Pauline Brice; John Radford; Vincent Ribrag; Daniel Molin; Theodoros P. Vassilakopoulos; et al.Akihiro TomitaBastian Von TresckowMargaret A. ShippYinghua ZhangAlejandro D. RicartArun BalakumaranCraig H. Moskowitz Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. Journal of Clinical Oncology 2017, 35, 2125-2132, 10.1200/jco.2016.72.1316.
- John Kuruvilla; Radhakrishnan Ramchandren; Armando Santoro; Ewa Paszkiewicz-Kozik; Robin Gasiorowski; Nathalie Johnson; Vladimir Melnichenko; Laura Maria Fogliatto; Iara Goncalves; Jose De Oliveira; et al.Valeria BuccheriGuilherme Fleury PeriniNeta GoldschmidtSergey AlekseevIryna KryachokNaohiro SekiguchiYing ZhuAkash NaharPatricia MarinelloPier Luigi Zinzani KEYNOTE-204: Randomized, open-label, phase III study of pembrolizumab (pembro) versus brentuximab vedotin (BV) in relapsed or refractory classic Hodgkin lymphoma (R/R cHL).. Journal of Clinical Oncology 2020, 38, 8005-8005, 10.1200/jco.2020.38.15_suppl.8005.
- Dennis A. Eichenauer; Andreas Engert; Nodular lymphocyte-predominant Hodgkin lymphoma: a unique disease deserving unique management. Hematology 2017, 2017, 324-328, 10.1182/asheducation-2017.1.324.
- Kerry J. Savage; Brian Skinnider; Mubarak Al-Mansour; Laurie H. Sehn; Randy D. Gascoyne; Joseph M. Connors; Treating limited-stage nodular lymphocyte predominant Hodgkin lymphoma similarly to classical Hodgkin lymphoma with ABVD may improve outcome. Blood 2011, 118, 4585-4590, 10.1182/blood-2011-07-365932.
- G. Enblad; B. Glimelius; C. Sundström; Treatment outcome in Hodgkin's disease in patients above the age of 60: A population-based study. Annals of Oncology 1991, 2, 297-302, 10.1093/oxfordjournals.annonc.a057939.
- Sven Borchmann; Andreas Engert; Boris Böll; Hodgkin lymphoma in elderly patients. Current Opinion in Oncology 2018, 30, 308-316, 10.1097/cco.0000000000000464.
- Boris Böll; Helen Görgen; Michael Fuchs; Annette Pluetschow; Hans Theodor Eich; Mario J. Bargetzi; Eckhart Weidmann; Christian Junghanß; Richard Greil; Alexander Scherpe; et al.Oliver SchmalzDennis A. EichenauerBastian Von TresckowAchim RotheVolker DiehlAndreas EngertPeter Borchmann ABVD in Older Patients With Early-Stage Hodgkin Lymphoma Treated Within the German Hodgkin Study Group HD10 and HD11 Trials. Journal of Clinical Oncology 2013, 31, 1522-1529, 10.1200/jco.2012.45.4181.
- Diana Wongso; Michael Fuchs; Annette Plütschow; Beate Klimm; Stephanie Sasse; Bernd Hertenstein; Georg Maschmeyer; Tom Vieler; Ulrich Dührsen; Walter Lindemann; et al.Walter AulitzkyVolker DiehlPeter BorchmannAndreas Engert Treatment-Related Mortality in Patients With Advanced-Stage Hodgkin Lymphoma: An Analysis of the German Hodgkin Study Group. Journal of Clinical Oncology 2013, 31, 2819-2824, 10.1200/jco.2012.47.9774.
- Paul J Bröckelmann; Dipl.-Math. Helen Goergen; Ulrich Keller; Julia Meissner; Karolin Trautmann; Teresa V Halbsguth; Stephanie Sasse; Martin Sökler; Andrea Kerkhoff; Stephan Mathas; et al.Andreas HüttmannMatthias BormannAndreas ZimmermannMichael FuchsBastian Von TresckowChristian BauesAndreas RosenwaldWolfram KlapperCarsten KobePeter BorchmannAndreas Engert Efficacy and Safety of Nivolumab and AVD in Early-Stage Unfavorable Hodgkin Lymphoma: Extended Follow-up from the GHSG Phase II Nivahl Trial. Blood 2020, 136, 6-7, 10.1182/blood-2020-138413.


