Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Minyoung Youn | + 1366 word(s) | 1366 | 2021-06-28 09:11:56 | | | |
| 2 | Vivi Li | Meta information modification | 1366 | 2021-07-07 08:45:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Youn, M. RSK Isoforms in AML. Encyclopedia. Available online: https://encyclopedia.pub/entry/11757 (accessed on 26 June 2026).
Youn M. RSK Isoforms in AML. Encyclopedia. Available at: https://encyclopedia.pub/entry/11757. Accessed June 26, 2026.
Youn, Minyoung. "RSK Isoforms in AML" Encyclopedia, https://encyclopedia.pub/entry/11757 (accessed June 26, 2026).
Youn, M. (2021, July 07). RSK Isoforms in AML. In Encyclopedia. https://encyclopedia.pub/entry/11757
Youn, Minyoung. "RSK Isoforms in AML." Encyclopedia. Web. 07 July, 2021.
Copy Citation
Ribosomal S6 Kinases (RSKs) are a group of serine/threonine kinases that function downstream of the Ras/Raf/MEK/ERK signaling pathway. Four RSK isoforms are directly activated by ERK1/2 in response to extracellular stimuli including growth factors, hormones, and chemokines. RSKs phosphorylate many cytosolic and nuclear targets resulting in the regulation of diverse cellular processes such as cell proliferation, survival, and motility. In hematological malignancies such as acute myeloid leukemia (AML), RSK isoforms are highly expressed and aberrantly activated resulting in poor outcomes and resistance to chemotherapy. Therefore, understanding RSK function in leukemia could lead to promising therapeutic strategies.
RSK isoforms
cancer
hematological malignancy
AML
RSK inhibitors
1. Introduction
The Ras-mitogen-activated protein kinase (MAPK) pathway is involved in the regulation of normal cell proliferation, survival, growth, and differentiation [1][2]. More than 30% of all human cancers are associated with abnormal control of this signaling network, resulting in gain of function and subsequent extracellular signal-regulated kinase (ERK) hyperactivation [3]. The Ras-MAPK pathway is initiated by a ligand binding to the receptor tyrosine kinase (RTK) receptor, followed by docking adaptor proteins such as growth factor receptor-bound protein 2 (GRB2) and Son of Sevenless (SOS), leading to activation of the associated Rat sarcoma (Ras) and recruitment of Raf. Raf then activates downstream mitogen-activated protein kinases kinase (MEK1/2) and ERK1/2 (Figure 1) [2].
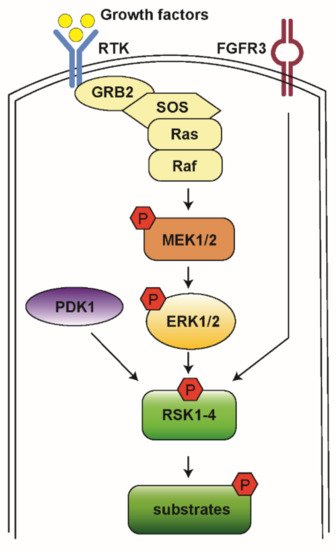
Figure 1. A schematic model for RSK activation. When RTK is stimulated by growth factor, it activates the docking proteins GRB2 and SOS. SOS triggers Ras to exchange guanosine diphosphate (GDP) to guanosine triphosphate (GTP) and then to become activated. Ras activates Raf kinases, which phosphorylate MEK1/2, ERK1/2, and RSK1-4. Then, RSKs phosphorylate various downstream substrates to mediate diverse cellular processes.
RSKs are a group of serine/threonine kinases that function in the MAPK signaling cascade and are the direct downstream effectors of ERK1/2. Four RSK isoforms are directly activated by ERK1/2 in response to extracellular stimuli including growth factors, hormones, and chemokines [4][5]. RSKs phosphorylate many cytosolic and nuclear targets resulting in the regulation of diverse cellular processes such as cell proliferation, survival, and motility. Therefore, the RSK isoforms represent attractive therapeutic targets for cancer [6][7]. Here we review the general roles of RSK isoforms and discuss their potential roles in AML and current pharmacological tools to inhibit their function.
2. RSK Isoforms in AML
AML is a genetically and phenotypically heterogeneous hematological malignancy characterized by the accumulation of immature myeloid blasts with peripheral blood cytopenia [8][9]. AML patients have an overall survival of less than 65% in children and 40% in adults. The current treatment options, including intensive chemotherapy and stem cell transplantation, are associated with significant morbidity and mortality [10]. Thus, it is critical to develop more effective and less toxic therapies for AML.
The Ras/Raf/MEK/ERK pathway has been reported to be constitutively activated in more than 50% of AML and acute lymphocytic leukemia (ALL) cases [11][12]. Thus, RSK isoforms play an important role in AML pathogenesis and progression. In addition, it has been observed that RSK1/2 are the predominant isoforms expressed in AML cells, whereas RSK4 has shown significantly lower expression in AML patients compared to healthy people. This suggests that downregulated RSK4 expression may lead to leukemia or negatively affect the prognosis of patients with AML [13]. We discuss below the roles of RSKs in AML pathogenesis (Figure 2) and how RSKs could be a therapeutic target for AML treatment.
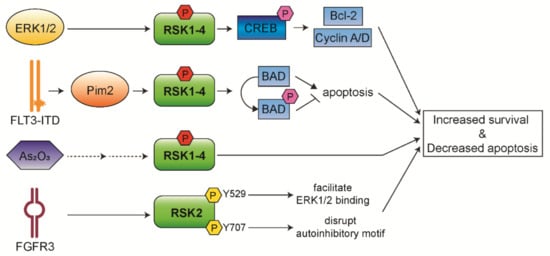
Figure 2. Functional mechanisms of RSK isoforms in AML. RSKs control survival and apoptosis of leukemic cells through aberrantly activated upstream signaling.
2.1. RSKs and CREB in AML Cell Survival
Our group has previously observed that approximately 60% of AML patients express CREB at high levels, and this is associated with an increased risk of relapse and decreased event-free survival [14][15]. Similarly, the expression and phosphorylation levels of RSKs are significantly increased in pediatric AML patients associated with poor survival [16][17]. Furthermore, we have observed that RSKs phosphorylate CREB on Ser133, and then phosphorylated CREB mediates proliferation and survival of myeloid cells through induced expression of Bcl-2, cyclin A, and cyclin D [14][16]. In addition, we have recently established that RSK inhibition inhibits AML cell proliferation through the regulation of mitotic exit [17]. A potent RSK inhibitor, BI-D1870, increases metaphase arrest by preventing the metaphase/anaphase transition, followed by induced apoptosis of AML patient cells through impeded association of cell division cycle 20 (CDC20) with anaphase promoting complex/cyclosome (APC/C) and increased mitotic arrest deficient 1 (MAD2) and CDC20 binding. Moreover, BI-D1870 treatment potentiates the anti-leukemic activity of vincristine via synergistically increased mitotic arrest and apoptosis in AML cells. Therefore, our findings suggest a novel therapy that overcomes vincristine resistance to AML cells.
2.2. RSKs in FLT3-ITD+ Cell Survival
FLT3 is a receptor-tyrosine kinase expressed on hematopoietic progenitor cells and plays an important role in proliferation, survival, and differentiation of these cells [18][19]. The internal tandem duplication mutation in FLT3 (FLT3-ITD) is the most frequent mutation in AML found in 25–30% of cases and associated with a poor prognosis [20]. FLT3-ITD leads to constitutive activation of the Ras/Raf/MEK/ERK pathway [11][12]. As a downstream regulator of this pathway, RSKs have essential roles in the pathogenesis and myeloid lineage determination of FLT3/ITD-induced hematopoietic transformation. Activated RSK1 phosphorylates and inactivates pro-apoptotic BAD protein, preventing the apoptosis of BaF/FLT3-ITD cells [21]. Inhibition of RSK1 expression reduces BAD phosphorylation resulting in the induced apoptosis of MV4-11, a cell line harboring FLT3-ITD. Targeting RSK2 by FMK, a RSK inhibitor, attenuates cell viability and induces significant apoptosis in human primary FLT3-ITD+ leukemic cells [22]. These findings suggest that combined inhibition of FLT3 and RSKs may be a viable therapeutic strategy to cure AML patients with FLT3-ITD.
In addition, recent studies have reported that RSKs have pro-survival functions as a new target of Pim2 kinase in relapsed FLT3-ITD+ AML cells [23][24]. Pim2 is a downstream target of FLT3-ITD+ AML cells and directly contributes to FLT3 inhibitor resistance [24]. Ectopic expression of RSK2 rescues the viability of Pim2-depleted cells through the regulation of Bax expression. These support the involvement of RSK2 in AML cell survival as a downstream of Pim2 and a novel therapeutic strategy against therapy-resistant FLT3-ITD+ AML.
Furthermore, it has been reported that FLT3-ITD activates RSK1 to enhance proliferation and survival of AML cells by activating mTORC1 and eIF4 [25]. Activated RSK1/2 via FLT3-ITD and MEK/ERK pathways phosphorylate TSC2 and eIF4B in cooperation with Pim2, thus activating mTORC1/S6K/4EBP1 pathway resulting in enhanced proliferation.
2.3. Alternative RSK Isoform Activation in FGFR3-Activated Cells
It has been shown that RSK2 has a role in hematopoietic transformation of AML and multiple myeloma via an alternative mechanism of RSK activation [26][27]. FGFR3 directly phosphorylates Tyr529 on RSK2, which facilitates inactive ERK1/2 binding to RSK2, and consequently phosphorylates and activates RSK2 [26]. FGFR3 additionally phosphorylates Tyr707 on RSK2 that may disrupt the auto-inhibitory αL-helix motif on C-terminal [27]. Targeted inhibition of RSK2 effectively induced apoptosis in FGFR3-expressing myeloma cells, suggesting that RSK2 is a critical signaling effector in FGFR3-mediated hematopoietic transformation.
2.4. RSKs in the Resistance to As2O3 Treatment
Arsenic trioxide (As2O3) treatment is an effective therapy for acute promyelocytic leukemia (APL) which is a subtype of AML, but shows no significant clinical activity in other non-APL subtype refractory or relapsed AML cases [28]. Recent studies have implicated that RSK1 is involved in the resistance of AML to As2O3 [29][30]. RSK1 is phosphorylated and activated during As2O3 treatment in different AML cell lines. This suggests that RSK1 counteracts As2O3-dependent anti-leukemic response by being activated in a negative feedback regulatory manner. Combined treatment of RSK1 inhibitor with As2O3 was found to result in more potent suppression of leukemic cells. These results suggest that RSK1 is a potentially important target to enhance the anti-leukemic properties of As2O3.
2.5. RSKs with Inhibition of SHH Signaling
Sonic Hedgehog (SHH) signaling is implicated in drug resistance for a range of human cancers [31]. A recent study showed that RSK inhibition overcomes resistance to inhibition of the SHH pathway in pediatric medulloblastoma [32]. In addition, SHH signaling has been found to play a role in the self-renewal of leukemia stem cells (LSCs) for CML and multiple myeloma [33][34][35]. These findings suggest that the combination of RSK and SHH inhibitors may be a complementary anti-leukemia strategy especially through regulating LSCs.
References
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362.
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83.
- Hoshino, R.; Chatani, Y.; Yamori, T.; Tsuruo, T.; Oka, H.; Yoshida, O.; Shimada, Y.; Ari-i, S.; Wada, H.; Fujimoto, J.; et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 1999, 18, 813–822.
- Romeo, Y.; Zhang, X.; Roux, P.P. Regulation and function of the RSK family of protein kinases. Biochem. J. 2012, 441, 553–569.
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758.
- Romeo, Y.; Roux, P.P. Paving the way for targeting RSK in cancer. Expert Opin. Ther. Targets. 2011, 15, 5–9.
- Sulzmaier, F.J.; Ramos, J.W. RSK isoforms in cancer cell invasion and metastasis. Cancer Res. 2013, 73, 6099–6105.
- Kavanagh, S.; Murphy, T.; Law, A.; Yehudai, D.; Ho, J.M.; Chan, S.; Schimmer, A.D. Emerging therapies for acute myeloid leukemia: Translating biology into the clinic. JCI Insight 2017, 2, e95679.
- Saygin, C.; Carraway, H.E. Emerging therapies for acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 93.
- Dombret, H.; Gardin, C. An update of current treatments for adult acute myeloid leukemia. Blood 2016, 127, 53–61.
- Ricciardi, M.R.; McQueen, T.; Chism, D.; Milella, M.; Estey, E.; Kaldjian, E.; Sebolt-Leopold, J.; Konopleva, M.; Andreeff, M. Quantitative single cell determination of ERK phosphorylation and regulation in relapsed and refractory primary acute myeloid leukemia. Leukemia 2005, 19, 1543–1549.
- Kornblau, S.M.; Womble, M.; Qiu, Y.H.; Jackson, C.E.; Chen, W.; Konopleva, M.; Estey, E.H.; Andreeff, M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 2006, 108, 2358–2365.
- Rafiee, M.; Keramati, M.R.; Ayatollahi, H.; Sadeghian, M.H.; Barzegar, M.; Asgharzadeh, A.; Alinejad, M. Down-Regulation of Ribosomal S6 kinase RPS6KA6 in Acute Myeloid Leukemia Patients. Cell J. 2016, 18, 159–164.
- Shankar, D.B.; Cheng, J.C.; Kinjo, K.; Federman, N.; Moore, T.B.; Gill, A.; Rao, N.P.; Landaw, E.M.; Sakamoto, K.M. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell. 2005, 7, 351–362.
- Cho, E.C.; Mitton, B.; Sakamoto, K.M. CREB and leukemogenesis. Crit. Rev. Oncog. 2011, 16, 37–46.
- Kwon, E.M.; Raines, M.A.; Blenis, J.; Sakamoto, K.M. Granulocyte-macrophage colony-stimulating factor stimulation results in phosphorylation of cAMP response element-binding protein through activation of pp90RSK. Blood 2000, 95, 2552–2558.
- Chae, H.D.; Dutta, R.; Tiu, B.; Hoff, F.W.; Accordi, B.; Serafin, V.; Youn, M.; Huang, M.; Sumarsono, N.; Davis, K.L.; et al. RSK inhibitor BI-D1870 inhibits acute myeloid leukemia cell proliferation by targeting mitotic exit. Oncotarget 2020, 11, 2387–2403.
- Hospital, M.A.; Green, A.S.; Maciel, T.T.; Moura, I.C.; Leung, A.Y.; Bouscary, D.; Tamburini, J. FLT3 inhibitors: Clinical potential in acute myeloid leukemia. Onco Targets Ther. 2017, 10, 607–615.
- Meshinchi, S.; Appelbaum, F.R. Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin. Cancer Res. 2009, 15, 4263–4269.
- Levis, M.; Small, D. FLT3: ITDoes matter in leukemia. Leukemia 2003, 17, 1738–1752.
- Yang, X.; Liu, L.; Sternberg, D.; Tang, L.; Galinsky, I.; DeAngelo, D.; Stone, R. The FLT3 internal tandem duplication mutation prevents apoptosis in interleukin-3-deprived BaF3 cells due to protein kinase A and ribosomal S6 kinase 1-mediated BAD phosphorylation at serine 112. Cancer Res. 2005, 65, 7338–7347.
- Elf, S.; Blevins, D.; Jin, L.; Chung, T.W.; Williams, I.R.; Lee, B.H.; Lin, J.X.; Leonard, W.J.; Taunton, J.; Khoury, H.J.; et al. p90RSK2 is essential for FLT3-ITD- but dispensable for BCR-ABL-induced myeloid leukemia. Blood. 2011, 117, 6885–6894.
- Hospital, M.A.; Jacquel, A.; Mazed, F.; Saland, E.; Larrue, C.; Mondesir, J.; Birsen, R.; Green, A.S.; Lambert, M.; Sujobert, P.; et al. RSK2 is a new Pim2 target with pro-survival functions in FLT3-ITD-positive acute myeloid leukemia. Leukemia 2018, 32, 597–605.
- Green, A.S.; Maciel, T.T.; Hospital, M.A.; Yin, C.; Mazed, F.; Townsend, E.C.; Pilorge, S.; Lambert, M.; Paubelle, E.; Jacquel, A.; et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci. Adv. 2015, 1, e1500221.
- Watanabe, D.; Nogami, A.; Okada, K.; Akiyama, H.; Umezawa, Y.; Miura, O. FLT3-ITD Activates RSK1 to Enhance Proliferation and Survival of AML Cells by Activating mTORC1 and eIF4B Cooperatively with PIM or PI3K and by Inhibiting Bad and BIM. Cancers 2019, 11, 1827.
- Kang, S.; Dong, S.; Gu, T.L.; Guo, A.; Cohen, M.S.; Lonial, S.; Khoury, H.J.; Fabbro, D.; Gilliland, D.G.; Bergsagel, P.L.; et al. FGFR3 activates RSK2 to mediate hematopoietic transformation through tyrosine phosphorylation of RSK2 and activation of the MEK/ERK pathway. Cancer Cell. 2007, 12, 201–214.
- Kang, S.; Elf, S.; Dong, S.; Hitosugi, T.; Lythgoe, K.; Guo, A.; Ruan, H.; Lonial, S.; Khoury, H.J.; Williams, I.R.; et al. Fibroblast growth factor receptor 3 associates with and tyrosine phosphorylates p90 RSK2, leading to RSK2 activation that mediates hematopoietic transformation. Mol. Cell Biol. 2009, 29, 2105–2117.
- Parmar, S.; Rundhaugen, L.M.; Boehlke, L.; Riley, M.; Nabhan, C.; Raji, A.; Frater, J.L.; Tallman, M.S. Phase II trial of arsenic trioxide in relapsed and refractory acute myeloid leukemia, secondary leukemia and/or newly diagnosed patients at least 65 years old. Leuk. Res. 2004, 28, 909–919.
- Galvin, J.P.; Altman, J.K.; Szilard, A.; Goussetis, D.J.; Vakana, E.; Sassano, A.; Platanias, L.C. Regulation of the kinase RSK1 by arsenic trioxide and generation of antileukemic responses. Cancer Biol. Ther. 2013, 14, 411–416.
- Beauchamp, E.M.; Kosciuczuk, E.M.; Serrano, R.; Nanavati, D.; Swindell, E.P.; Viollet, B.; O’Halloran, T.V.; Altman, J.K.; Platanias, L.C. Direct binding of arsenic trioxide to AMPK and generation of inhibitory effects on acute myeloid leukemia precursors. Mol. Cancer Ther. 2015, 14, 202–212.
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354.
- Pambid, M.R.; Berns, R.; Adomat, H.H.; Hu, K.; Triscott, J.; Maurer, N.; Zisman, N.; Ramaswamy, V.; Hawkins, C.E.; Taylor, M.D.; et al. Overcoming resistance to Sonic Hedgehog inhibition by targeting p90 ribosomal S6 kinase in pediatric medulloblastoma. Pediatr. Blood Cancer. 2014, 61, 107–115.
- Lin, T.L.; Levy, M.Y. Acute myeloid leukemia: Focus on novel therapeutic strategies. Clin. Med. Insights Oncol. 2012, 6, 205–217.
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779.
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kin, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053.
More
Information
Subjects:
Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
07 Jul 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No