 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lung-Yi Loey Mak | + 5404 word(s) | 5404 | 2021-06-21 04:29:16 | | | |
| 2 | Bruce Ren | -21 word(s) | 5383 | 2021-07-06 04:04:40 | | |
Video Upload Options
Globally, chronic hepatitis B (CHB) infection is one of the leading causes of liver failure, decompensated cirrhosis, and hepatocellular carcinoma. Existing antiviral therapy can suppress viral replication but not fully eradicate the virus nor the risk of liver-related complications. Novel treatments targeting alternative steps of the viral cycle or to intensify/restore the host’s immunity are being developed. We discuss novel drugs that have already entered clinical phases of development. Agents that interfere with specific steps of HBV replication include RNA interference, core protein allosteric modulation, and inhibition of viral entry or viral protein excretion (NAPs and STOPS). Agents that target the host’s immunity include toll-like receptor agonists, therapeutic vaccines, immune checkpoint modulators, soluble T-cell receptors, and monoclonal antibodies. Most have demonstrated favorable results in suppression of viral proteins and genomic materials (i.e., HBV DNA and/or pre-genomic RNA), and/or evidence on host-immunity restoration including cytokine responses and T-cell activation. Given the abundant clinical experience and real-world safety data with the currently existing therapy, any novel agent for CHB should be accompanied by convincing safety data. Combination therapy of nucleos(t)ide analogue, a novel virus-directing agent, and/or an immunomodulatory agent will be the likely approach to optimize the chance of a functional cure in CHB.
1. Introduction
2. Functional Cure: The Preferred Treatment Endpoint
| Therapeutic Outcome | Blood | Liver | ||||
|---|---|---|---|---|---|---|
| HBV DNA | HBsAg | Anti-HBs * | Anti-HBc | cccDNA | Integrated DNA | |
| Partial cure | − | + | − | −/+ | + | + |
| Functional cure | − | − | −/+ | −/+ | + | + |
| Complete cure | − | − | −/+ | −/+ | − | + |
| Sterilizing cure | − | − | −/+ | −/+ | − | − |
3. Novel Therapeutic Approaches
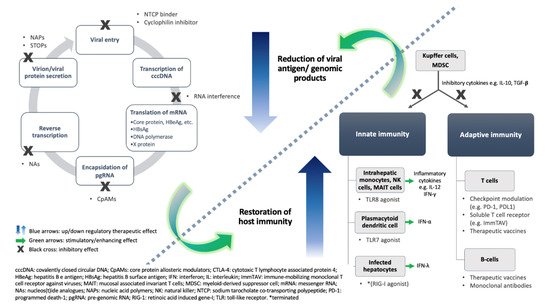
3.1. Virus-Directing Agents
| Main Mechanism | Remarks | Drug Names | Delivery | Phase | Clinical Trial Identifier | |
|---|---|---|---|---|---|---|
| Inhibition of viral entry | NTCP binding | Myrcludex B/Bulevertide | SC | 3 | NCT03852719 | |
| Cyclophilin inhibitor | CRV-431 | Oral | 1 | NCT03596697 | ||
| RNA interference | siRNA | Dicerna GAIXc-HBVS (RG 6346) | SC | 1/2 | NCT03772249 | |
| JNJ 3989 (ARO-HBV 1 & ARO-HBV 2) | 2 | NCT04129554 | ||||
| AB-729 | 2 | NCT04820686 | ||||
| VIR-2218 (ALN-HBV) | 2 | NCT03672188 | ||||
| ASO | GSK-836 (ISIS-358)-non GaINAc | SC | 2 | NCT04449029 | ||
| GSK-404-GaiNAc | 2 | NCT03020745 | ||||
| RO7062931-GaiNAc | 1 | NCT03038113 | ||||
| Inhibition of capsid formation | CpAM | Class 1 | GLS-4 (Morphothiadin)/ritonavir | Oral | 2 | NCT04147208 |
| Class 2 | ABI-HB0731 (Vebicorvir) | 2 | NCT03780543 | |||
| Class 2 | ABI-H2158 | 2 | NCT04398134 | |||
| Class 2 | JNJ-6379 | 2 | NCT03361956 | |||
| Class 2 | EDP-514 | 1 | NCT04470388 NCT04008004 |
|||
| Not disclosed | QL-007 | 1 | NCT03770624 NCT03244085 |
|||
| Class 2 | ZM-H1505R | 1 | NCT04220801 | |||
| Class 2 | ABI-H3733 | 1 | NCT04271592 | |||
| Class 2 | ALG-000184 (prodrug of ALG-001075) | 1 | NCT04536337 | |||
| Class 1 | RO7049389 (RG7907) | 1 | NCT02952924 | |||
| Inhibition of HBsAg release | Nucleic acid polymer | REP 2139 or REP 2165 | IV | 2 | NCT02565719 | |
| STOPS | ALG-010133 | SC | 1 | NCT04485663 | ||
| Interaction with host nuclear receptor | FXR agonist | EYP001 | Oral | 2 | NCT04465916 | |
| Enhancement of innate/adaptive immunity | TLR agonist | RO7020531 (also known as RG-7854, TLR7) | Oral | 1 | NCT02956850 | |
| Vesatolimod (TLR7, GS-9620) | 2 | NCT02166047 | ||||
| Selgantolimod (TLR8, GS-9688) | 2 | NCT03491553 | ||||
| T cell | ASC22 (Anti-PDL1) | SC | 2 | NCT04465890 | ||
| Cemiplimab (Anti-PD1) | IV | 1/2 | NCT04046107 | |||
| Nivolumab (Anti-PD1) | IV | 1 | ACTRN12615001133527 * | |||
| APG-1387 (apoptosis inducer) | IV | 2 | NCT04568265 | |||
| IMC-I109V (soluble T-cell receptor, ImmTAV molecule) | IV | 1/2 | NCT03973333 | |||
| Therapeutic vaccine | HeberNasvac (ABX-203) | Intranasal | 3 | NCT02249988 | ||
| GS-4774 | SC | 2 | NCT01943799 | |||
| HepTcell | IM | 2 | NCT04684914 | |||
| AIC649 | IV | 1 | Not applicable | |||
| HB-110 | EP | 1 | NCT01641536 | |||
| VTP-300 | IM | 1/2 | NCT04778904 | |||
| JNJ 64300535 | EP | 1 | NCT03463369 | |||
| BRII-179 (VBI-2601) | IM | 1/2 | NCT04749368 | |||
| TG-1050 | SC | 1 | NCT02428400 | |||
| INO-1800 | EP | 1 | NCT02431312 | |||
| Monoclonal antibody | GC1102 | IV | 2 | NCT03801798 | ||
| VIR-3434 | SC/IV | 1 | NCT04423393 | |||
3.1.1. Inhibition of Viral Entry
3.1.2. Interference of RNA
3.1.3. Inhibition of Functional Capsid Assembly or Encapsidation
3.1.4. Inhibition of Viral Protein Export
3.1.5. Farnesoid X Receptor Agonist
3.2. Enhancement of Host Immunity
4. Combination Strategies
With the efficacy and safety data of the individual novel agents mentioned above, the approach of combining two or more novel agents to reduce viral antigen load, inhibit viral replication, and stimulate immunity is being actively explored.
The triple combination of RNAi (monthly injections for three doses of JNJ-3989) + CpAM (daily oral doses of JNJ-6379 for 85 days) + NA (daily oral doses beyond the end of CpAM dosing) led to mean 1.7 logs reduction in HBsAg on day 113 in 12 CHB patients. In addition, other viral products including HBV DNA, HBV RNA, and HBcrAg were profoundly suppressed. This combination therapy was in general well-tolerated with no serious or severe adverse events reported. Mild ALT flares were observed in five patients and were attributed to therapeutic flares [89].
A few more combination therapies are currently being evaluated in a phase 2 study that evaluates the safety and efficacy of multiple combination therapies (ClinicalTrials.gov identifier: NCT04225715). One of them is a similar example as the above with triple combination with RNAi (RG6346) + CpAM (RO7049389) + NA. Other examples include: triple combination of RNAi (RG6346) + TLR7 agonist (RO7020531) + NA, triple combination with RNAi (RG6346) + PEG-IFN + NUC, and triple combination with CpAM (RO7049389) + TLR agonist (RO7020531) + NA. The approach of sequential use of therapeutic vaccines after antigen knockdown by RNAi has been explored in mice models that showed encouraging results in suppression of viral burden and stimulation in the number of functional HBV-specific T cells and production of HBV-neutralizing antibodies [90]. Clinical studies using this approach are awaited.
Some agents will require combination with NAs, especially CpAMs, NAPs, and most immune modulatory agents, in contrast to RNAi. This is likely to be related to whether the antiviral effect of the individual agent leads to silencing of cccDNA transcriptional activity, which is the case for RNAi via post-transcriptional knockdown of HBV transcripts. Theoretically, sequential combinational therapy will result in rapid decline in HBsAg (by virus-directing agents) which is beneficial for subsequent immune stimulation therapy [91]. In transgenic mice models, GaINac-conjugated siRNA followed by therapeutic vaccine showed significantly stronger immune stimulatory effect in terms of development of polyfunctional, HBV-specific CD8+ T cells compared to mice given control RNAs with NA, and therapeutic vaccine [90]. Although many RNAi-based combination therapies are still tested in clinical trials in combination with NAs, it remains to be unveiled whether the viral suppressive effects of NA can be substituted by other virus-directing agents.
5. Conclusions
The currently preferred treatment endpoint for CHB is functional cure. Novel agents work on different steps of viral replication or the host’s immune system, in order to reduce viral burden, inhibit viral replication, and restore host immunity. The therapeutic effects of both virus-directed and host-immunity-directed agents are intercalated and are equally crucial for achieving a functional cure. Most agents currently in clinical phases of development demonstrated favorable results in suppression of viral proteins and genomic materials, with initially promising results of enhancing the functional cure of CHB. With an increasing number of novel agents entering clinical phases of development, it is expected that each agent should demonstrate favorable safety data, and long-term follow-up data in the subjects that participated in these trials will be an important consideration. Most trials aimed to achieve broad eligibility by recruiting CHB patients with variable duration of infection and viral activity. However, it is likely that specific subgroups of patients will benefit most from selected agents so that a finite duration of therapy (partial cure or functional cure if HBsAg seroclearance can be achieved) instead of lifelong NA treatment can become feasible. For all trials mentioned, no cirrhotic patients were included and this will need to be addressed when more safety data are available. Combination therapy with two or more novel agents theoretically leads to synergistic therapeutic effects. NA and/or PEG-IFN are still the backbone of these combination regimens. The best cocktail of therapies is being sought, and different regimens are likely to be needed for different patient populations with various viral factors (e.g., HBsAg levels, previous treatment exposure), and host factors (e.g., HLA allele, gender).
References
- Razavi-Shearer, D.; Gamkrelidze, I.; Nguyen, M.H.; Chen, D.-S.; Van Damme, P.; Abbas, Z.; Abdulla, M.; Abou Rached, A.; Adda, D.; Aho, I.; et al. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403.
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398.
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018, 67, 1560–1599.
- Lin, S.M.; Sheen, I.S.; Chien, R.N.; Chu, C.M.; Liaw, Y.F. Long-term beneficial effect of interferon therapy in patients with chronic hepatitis B virus infection. Hepatology 1999, 29, 971–975.
- Seto, W.K.; Lau, E.H.; Wu, J.T.; Hung, I.F.; Leung, W.K.; Cheung, K.S.; Fung, J.; Lai, C.L.; Yuen, M.F. Effects of nucleoside analogue prescription for hepatitis B on the incidence of liver cancer in Hong Kong: A territory-wide ecological study. Aliment. Pharmacol Ther. 2017, 45, 501–509.
- Thiele, M.; Gluud, L.L.; Dahl, E.K.; Krag, A. Antiviral therapy for prevention of hepatocellular carcinoma and mortality in chronic hepatitis B: Systematic review and meta-analysis. BMJ Open 2013, 3, e003265.
- Seto, W.K.; Hui, A.J.; Wong, V.W.; Wong, G.L.; Liu, K.S.; Lai, C.L.; Yuen, M.F.; Chan, H.L. Treatment cessation of entecavir in Asian patients with hepatitis B e antigen negative chronic hepatitis B: A multicentre prospective study. Gut 2015, 64, 667–672.
- Lai, C.L.; Wong, D.K.; Wong, G.T.; Seto, W.K.; Fung, J.; Yuen, M.F. Rebound of HBV DNA after cessation of nucleos/tide analogues in chronic hepatitis B patients with undetectable covalently closed. JHEP Rep. 2020, 2, 100112.
- Mak, L.Y.; Seto, W.K.; Lai, C.L.; Yuen, M.F. DNA polymerase inhibitors for treating hepatitis B: A safety evaluation. Expert Opin. Drug Saf. 2016, 15, 383–392.
- Lai, C.L.; Wong, D.; Ip, P.; Kopaniszen, M.; Seto, W.K.; Fung, J.; Huang, F.Y.; Lee, B.; Cullaro, G.; Chong, C.K.; et al. Reduction of covalently closed circular DNA with long-term nucleos(t)ide analogue treatment in chronic hepatitis B. J. Hepatol. 2017, 66, 275–281.
- Cornberg, M.; Lok, A.S.; Terrault, N.A.; Zoulim, F.; Berg, T.; Brunetto, M.R.; Buchholz, S.; Buti, M.; Chan, H.L.Y.; Chang, K.-M.; et al. Guidance for design and endpoints of clinical trials in chronic hepatitis B—Report from the 2019 EASL-AASLD HBV Treatment Endpoints Conference. Hepatology 2019, 72, 539–557.
- Yip, T.C.; Wong, G.L.; Wong, V.W.; Tse, Y.K.; Lui, G.C.; Lam, K.L.; Chan, H.L. Durability of hepatitis B surface antigen seroclearance in untreated and nucleos(t)ide analogue-treated patients. J. Hepatol. 2017, 68, 63–72.
- Seto, W.K.; Cheung, K.S.; Wong, D.K.; Huang, F.Y.; Fung, J.; Liu, K.S.; Lai, C.L.; Yuen, M.F. Hepatitis B surface antigen seroclearance during nucleoside analogue therapy: Surface antigen kinetics, outcomes, and durability. J. Gastroenterol. 2016, 51, 487–495.
- Yuen, M.F.; Wong, D.K.; Fung, J.; Ip, P.; But, D.; Hung, I.; Lau, K.; Yuen, J.C.; Lai, C.L. HBsAg Seroclearance in chronic hepatitis B in Asian patients: Replicative level and risk of hepatocellular carcinoma. Gastroenterology 2008, 135, 1192–1199.
- Yip, T.C.; Chan, H.L.; Wong, V.W.; Tse, Y.K.; Lam, K.L.; Wong, G.L. Impact of age and gender on risk of hepatocellular carcinoma after hepatitis B surface antigen seroclearance. J. Hepatol. 2017, 67, 902–908.
- Kim, G.A.; Lee, H.C.; Kim, M.J.; Ha, Y.; Park, E.J.; An, J.; Lee, D.; Shim, J.H.; Kim, K.M.; Lim, Y.S. Incidence of hepatocellular carcinoma after HBsAg seroclearance in chronic hepatitis B patients: A need for surveillance. J. Hepatol. 2015, 62, 1092–1099.
- Anderson, R.T.; Choi, H.S.J.; Lenz, O.; Peters, M.G.; Janssen, H.L.A.; Mishra, P.; Donaldson, E.; Westman, G.; Buchholz, S.; Miller, V.; et al. Association Between Seroclearance of Hepatitis B Surface Antigen and Long-Term Clinical Outcomes of Patients With Chronic Hepatitis B Virus Infection: Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2020, 19, 463–472.
- Kim, G.A.; Lim, Y.S.; An, J.; Lee, D.; Shim, J.H.; Kim, K.M.; Lee, H.C.; Chung, Y.H.; Lee, Y.S.; Suh, D.J. HBsAg seroclearance after nucleoside analogue therapy in patients with chronic hepatitis B: Clinical outcomes and durability. Gut 2014, 63, 1325–1332.
- Mak, L.Y.; Seto, W.K.; Hui, R.W.; Fung, J.; Wong, D.K.; Lai, C.L.; Yuen, M.F. Fibrosis evolution in chronic hepatitis B e antigen-negative patients across a 10-year interval. J. Viral Hepat. 2019, 26, 818–827.
- Berg, T.; Lampertico, P. The times they are a-changing—A refined proposal for finite HBV nucleos(t)ide analogue therapy. J. Hepatol. 2021.
- Tout, I.; Loureiro, D.; Mansouri, A.; Soumelis, V.; Boyer, N.; Asselah, T. Hepatitis B surface antigen seroclearance: Immune mechanisms, clinical impact, importance for drug development. J. Hepatol. 2020, 73, 409–422.
- Milich, D.R.; Jones, J.E.; Hughes, J.L.; Price, J.; Raney, A.K.; McLachlan, A. Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc. Natl. Acad. Sci. USA 1990, 87, 6599–6603.
- Jiang, M.; Broering, R.; Trippler, M.; Poggenpohl, L.; Fiedler, M.; Gerken, G.; Lu, M.; Schlaak, J.F. Toll-like receptor-mediated immune responses are attenuated in the presence of high levels of hepatitis B virus surface antigen. J. Viral Hepat. 2014, 21, 860–872.
- Rehermann, B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat. Med. 2013, 19, 859–868.
- Chen, M.; Sallberg, M.; Hughes, J.; Jones, J.; Guidotti, L.G.; Chisari, F.V.; Billaud, J.N.; Milich, D.R. Immune tolerance split between hepatitis B virus precore and core proteins. J. Virol. 2005, 79, 3016–3027.
- Maini, M.K.; Boni, C.; Ogg, G.S.; King, A.S.; Reignat, S.; Lee, C.K.; Larrubia, J.R.; Webster, G.J.; McMichael, A.J.; Ferrari, C.; et al. Direct ex vivo analysis of hepatitis B virus-specific CD8(+) T cells associated with the control of infection. Gastroenterology 1999, 117, 1386–1396.
- Penna, A.; Artini, M.; Cavalli, A.; Levrero, M.; Bertoletti, A.; Pilli, M.; Chisari, F.V.; Rehermann, B.; Del Prete, G.; Fiaccadori, F.; et al. Long-lasting memory T cell responses following self-limited acute hepatitis B. J. Clin. Investig. 1996, 98, 1185–1194.
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology 2010, 138, 682–693.
- Wongjitrat, C.; Sukwit, S.; Chuenchitra, T.; Seangjaruk, P.; Rojanasang, P.; Romputtan, P.; Srisurapanon, S. CTLA-4 and its ligands on the surface of T- and B-lymphocyte subsets in chronic hepatitis B virus infection. J. Med. Assoc. Thail. Chotmaihet Thangphaet 2013, 96, S54–S59.
- Burton, A.R.; Pallett, L.J.; McCoy, L.E.; Suveizdyte, K.; Amin, O.E.; Swadling, L.; Alberts, E.; Davidson, B.R.; Kennedy, P.T.; Gill, U.S.; et al. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J. Clin. Investig. 2018, 128, 4588–4603.
- Fang, Z.; Li, J.; Yu, X.; Zhang, D.; Ren, G.; Shi, B.; Wang, C.; Kosinska, A.D.; Wang, S.; Zhou, X.; et al. Polarization of Monocytic Myeloid-Derived Suppressor Cells by Hepatitis B Surface Antigen Is Mediated via ERK/IL-6/STAT3 Signaling Feedback and Restrains the Activation of T Cells in Chronic Hepatitis B Virus Infection. J. Immunol. 2015, 195, 4873–4883.
- Kondo, Y.; Ninomiya, M.; Kakazu, E.; Kimura, O.; Shimosegawa, T. Hepatitis B surface antigen could contribute to the immunopathogenesis of hepatitis B virus infection. ISRN Gastroenterol. 2013, 2013, 935295.
- Yeo, Y.H.; Ho, H.J.; Yang, H.I.; Tseng, T.C.; Hosaka, T.; Trinh, H.N.; Kwak, M.S.; Park, Y.M.; Fung, J.Y.Y.; Buti, M.; et al. Factors Associated With Rates of HBsAg Seroclearance in Adults With Chronic HBV Infection: A Systematic Review and Meta-analysis. Gastroenterology 2019, 156, 635–646.
- Yeo, Y.H.; Tseng, T.C.; Hosaka, T.; Cunningham, C.; Fung, J.Y.Y.; Ho, H.J.; Kwak, M.S.; Trinh, H.N.; Ungtrakul, T.; Yu, M.L.; et al. Incidence, Factors, and Patient-Level Data for Spontaneous HBsAg Seroclearance: A Cohort Study of 11,264 Patients. Clin. Transl. Gastroenterol. 2020, 11, e00196.
- Yuen, M.F.; Lai, C.L. Hepatitis B in 2014: HBV research moves forward–receptors and reactivation. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 70–72.
- Wedemeyer, H.; Schöneweis, K.; Bogomolov, P.O.; Chulanov, V.; Stepanova, T.; Viacheslav, M.; Allweiss, L.; Dandri, M.; Ciesek, S.; Dittmer, U.; et al. 48 weeks of high dose (10 mg) bulevirtide as monotherapy or with peginterferon alfa-2a in patients with chronic HBV/HDV co-infection. J. Hepatol. 2020, 73, S52–S53.
- Urban, S.; Bartenschlager, R.; Kubitz, R.; Zoulim, F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology 2014, 147, 48–64.
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J. Hepatol. 2016, 65, 490–498.
- Shimura, S.; Watashi, K.; Fukano, K.; Peel, M.; Sluder, A.; Kawai, F.; Iwamoto, M.; Tsukuda, S.; Takeuchi, J.S.; Miyake, T.; et al. Cyclosporin derivatives inhibit hepatitis B virus entry without interfering with NTCP transporter activity. J. Hepatol. 2017, 66, 685–692.
- Gallay, P.; Ure, D.; Bobardt, M.; Chatterji, U.; Ou, J.; Trepanier, D.; Foster, R. The cyclophilin inhibitor CRV431 inhibits liver HBV DNA and HBsAg in transgenic mice. PLoS ONE 2019, 14, e0217433.
- Agrawal, N.; Dasaradhi, P.V.; Mohmmed, A.; Malhotra, P.; Bhatnagar, R.K.; Mukherjee, S.K. RNA interference: Biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. MMBR 2003, 67, 657–685.
- Mello, C.C.; Conte, D., Jr. Revealing the world of RNA interference. Nature 2004, 431, 338–342.
- Watts, J.K.; Corey, D.R. Silencing disease genes in the laboratory and the clinic. J. Pathol. 2012, 226, 365–379.
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807.
- Veedu, R.N.; Wengel, J. Locked nucleic acid as a novel class of therapeutic agents. RNA Biol. 2009, 6, 321–323.
- Kauppinen, S.; Vester, B.; Wengel, J. Locked nucleic acid: High-affinity targeting of complementary RNA for RNomics. RNA Towards Med. 2006, 173, 405–422.
- Javanbakht, H.; Mueller, H.; Walther, J.; Zhou, X.; Lopez, A.; Pattupara, T.; Blaising, J.; Pedersen, L.; Albaek, N.; Jackerott, M.; et al. Liver-Targeted Anti-HBV Single-Stranded Oligonucleotides with Locked Nucleic Acid Potently Reduce HBV Gene Expression In Vivo. Mol. Ther. Nucleic Acids 2018, 11, 441–454.
- Gane, E.; Locarnini, S.; Lim, T.; Strasser, S.; Sievert, W.; Cheng, W.; Thompson, A.; Given, B.; Schluep, T.; Hamilton, J.; et al. Short-term treatment with RNA interference therapy, JNJ-3989, results in sustained hepatitis B surface antigen suppression in patients with chronic hepatitis B receiving nucleos(t)ide analogue treatment. In Proceedings of the Digital International Liver Congress, 27–29 August 2020; p. S20.
- Gane, E.; Lim, Y.; Tangkijvanich, P.; O’Beirne, J.; Lim, T.; Bakardjiev, A.; Ding, X.; Connolly, L.; Huang, S.; Kim, J.; et al. Preliminary safety and antiviral activity of VIR-2218, an X-targeting HBV RNAi therapeutic, in chronic hepatitis B patients. In Proceedings of the Digital International Liver Congress, 27–29 August 2020; pp. S50–S51.
- Yuen, M.; Lim, T.; Kin, W.; Tongkijvonich, P.; Yoon, J.; Sievert, W.; Sukeepoisornjoroen, W.; Thompson, A.; CSchwabe, C.; Brown, B.; et al. HBV RNAi inhibitor RG6346 in Phase 1b-2a trial was safe, well-tolerated, and resulted in substantial and durable reductions in serum HBsAg levels. In Proceedings of the Digital Liver Meeting, 11–16 November 2020.
- Yuen, M.; Berliba, E.; Kim, Y.; Holmes, J.; Lim, Y.; Strasser, S.; Schwabe, C.; Jucov, A.; Lee, A.; Thi, E.; et al. Safety and pharmacodynamics of the GalNAc-siRNA AB-729 in subjects with chronic hepatitis B infection. In Proceedings of the Digital Liver Meeting, 11–16 November 2020.
- Yuen, M.F.; Wong, D.K.; Schluep, T.; Lai, C.L.; Ferrari, C.; Locarnini, S.; Lo, R.C.; Gish, R.G.; Hamilton, J.; Wooddell, C.I.; et al. Long-term serological, virological and histological responses to RNA inhibition by ARC-520 in Chinese chronic hepatitis B patients on entecavir treatment. Gut 2021.
- Yuen, R.; Heo, J.; Jang, J.; Yoon, J.-H.; Kweon, Y.; Park, S.-J.; Bennett, F.; Kwoh, J. Hepatitis B virus (HBV) surface antigen (HBsAg) inhibition with isis 505358 in chronic hepatitis B (CHB) patients on stable nucleos(t)ide analogue (NA)-naïve CHB patients: Phase 2a, randomized, double-blind, placebo-controlled study. J. Hepatol. 2020, 73, S49–S50.
- Mak, L.Y.; Wong, D.K.; Seto, W.K.; Lai, C.L.; Yuen, M.F. Hepatitis B core protein as a therapeutic target. Expert Opin. Ther. Targets 2017, 21, 1153–1159.
- Yuen, M.F.; Schwabe, C.; Tanwandee, T.; Jin, Y.; Gao, L.; Zhou, X.; Das, S.; Wang, Y.; Lemenuel-Diot, A.; Cosson, A.; et al. RO7049389, a core protein allosteric modulator, demonstrates robust decline in HBV DNA and HBV RNA in chronic HBV infected patients. In Proceedings of the International Liver Congress, EASL2019, Vienna, Austria, 10–14 April 2019; p. e491.
- Zhang, M.; Zhang, J.; Tan, Y.; Xin, Y.; Gao, H.; Zheng, S.; Yi, Y.; Zhang, J.; Wu, C.; Zhao, Y.; et al. Efficacy and safety of GLS4/ritonavir combined with entecavir in HBeAg-positive patients with chronic hepatitis B: Interim results from phase 2b, multi-center study. J. Hepatol. 2020, 73 (Suppl. 1), S878–S879.
- Janssen, H.; Hou, J.; Asselah, T.; Chan, H.; Zoulim, F.; Tanaka, Y.; Janczewska, E.; Nahass, R.; Bourgeois, S.; Buti, M.; et al. Efficacy and Safety Results of the Phase 2 JNJ-56136379 JADE Study in Patients with Chronic Hepatitis B: Interim Week 24 Data. In Proceedings of the Digital ILC, 27–29 August 2020.
- Gane, E.; Sulkowski, M.; Ma, X.; Nguyen, T.; Hann, H.; Hassanein, T.; Elkhashab, M.; Nahass, R.; Chan, S.; Bennett, M.; et al. Viral response and safety following discontinuation of treatment with the core inhibitor vebicorvir and a nucleos(t)ide reverse transcriptase inhibitor in patients with HBeAg positive or negative chronic hepatitis B virus infection. J. Hepatol. 2021. In Press.
- Yuen, M.F.; Agarwal, K.; Gane, E.J.; Schwabe, C.; Ahn, S.H.; Kim, D.J.; Lim, Y.S.; Cheng, W.; Sievert, W.; Visvanathan, K.; et al. Safety, pharmacokinetics, and antiviral effects of ABI-H0731, a hepatitis B virus core inhibitor: A randomised, placebo-controlled phase 1 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 152–166.
- Jekle, A.; Zhang, Q.; Serebryany, V.; Welch, M.; Liu, J.; Vendeville, S.; Debing, Y.; Kum, D.B.; Ren, S.; Liu, C.; et al. Best-in-class preclinical characteristics of ALG-000184, a prodrug of the capsid assembly modulator ALG-001075 for the treatment of chronic hepatitis B. In Proceedings of the Digital Liver Meeting 2020, Boston, MA, USA, 13–16 November 2020.
- Boulon, R.; Blanchet, M.; Lemasson, M.; Vaillant, A.; Labonte, P. Characterization of the antiviral effects of REP 2139 on the HBV lifecycle in vitro. Antivir. Res. 2020, 183, 104853.
- Vaillant, A. REP 2139: Antiviral Mechanisms and Applications in Achieving Functional Control of HBV and HDV Infection. ACS Infect. Dis. 2019, 5, 675–687.
- Bazinet, M.; Pantea, V.; Placinta, G.; Moscalu, I.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Iarovoi, L.; Smesnoi, V.; Musteata, T.; et al. Safety and Efficacy of 48 Weeks REP 2139 or REP 2165, Tenofovir Disoproxil, and Pegylated Interferon Alfa-2a in Patients With Chronic HBV Infection Naive to Nucleos(t)ide Therapy. Gastroenterology 2020, 158, 2180–2194.
- Nie, Y.; Tan, H.; Kao, C.; Ren, S.; Pandey, R.; Chanda, S.; Lin, T.-I.; Blatt, L.M.; Beigelman, L.N.; Symons, J.A.; et al. ALG-010133, a Representative S-Antigen Transport-inhibiting Oligonucleotide Polymer (STOPSTM) Effectively Inhibits Hepatitis B Surface Antigen (HBsAg) Secretion in Multiple Hepatitis B Virus (HBV) Cell Models. In Proceedings of the Digital Liver Meeting, Boston, MA, USA, 13–16 November 2020.
- Maini, M.K.; Burton, A.R. Restoring, releasing or replacing adaptive immunity in chronic hepatitis B. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 662–675.
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165.
- Ramiere, C.; Scholtes, C.; Diaz, O.; Icard, V.; Perrin-Cocon, L.; Trabaud, M.A.; Lotteau, V.; Andre, P. Transactivation of the hepatitis B virus core promoter by the nuclear receptor FXRalpha. J. Virol. 2008, 82, 10832–10840.
- Mouzannar, K.; Fusil, F.; Lacombe, B.; Ollivier, A.; Menard, C.; Lotteau, V.; Cosset, F.L.; Ramiere, C.; Andre, P. Farnesoid X receptor-alpha is a proviral host factor for hepatitis B virus that is inhibited by ligands in vitro and in vivo. FASEB J. 2019, 33, 2472–2483.
- Erken, R.; Stelma, F.; Roy, E.; Diane, S.; Andre, P.; Vonderscher, J.; Eric, M.; Tim, S.; Philippe, P.; Christian, L.; et al. First clinical evaluation in chronic hepatitis B patients of the synthetic farnesoid X receptor agonist EYP001. J. Hepatol. 2018, 68, S488–S489.
- Janssen, H.L.A.; Brunetto, M.R.; Kim, Y.J.; Ferrari, C.; Massetto, B.; Nguyen, A.H.; Joshi, A.; Woo, J.; Lau, A.H.; Gaggar, A.; et al. Safety, efficacy and pharmacodynamics of vesatolimod (GS-9620) in virally suppressed patients with chronic hepatitis B. J. Hepatol. 2018, 68, 431–440.
- Gane, E.J.; Kim, H.J.; Visvanathan, K.; Kim, Y.J.; Nguyen, A.H.; Wallin, J.J.; Chen, D.Y.; McDonald, C.; Arora, P.; Tan, S.K.; et al. Safety, pharmacokinetics, and pharmacodynamics of the oral TLR8 agonist selgantolimod in chronic hepatitis B. Hepatology 2021.
- Gane, E.; Verdon, D.J.; Brooks, A.E.; Gaggar, A.; Nguyen, A.H.; Subramanian, G.M.; Schwabe, C.; Dunbar, P.R. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: A pilot study. J. Hepatol. 2019, 71, 900–907.
- Fergusson, J.R.; Wallace, Z.; Connolly, M.M.; Woon, A.P.; Suckling, R.J.; Hine, D.W.; Barber, C.; Bunjobpol, W.; Choi, B.S.; Crespillo, S.; et al. Immune-Mobilizing Monoclonal T Cell Receptors Mediate Specific and Rapid Elimination of Hepatitis B-Infected Cells. Hepatology 2020, 72, 1528–1540.
- Allele frequency net database (AFND) 2020 update: Gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2020, 48, D783–D788.
- Leonard, S.; Paterson, R.; Godinho, L.; Howe, D.; Monteiro, M.; Hague, R.M.; Atkin, K.; Sarkar, A.; Suckling, R.; Bunjobpol, W.; et al. NOVEL HLA-E SPECIFIC IMMTAV® MOLECULES FOR THE TREATMENT OF HEPATITIS B. In Proceedings of the Digital Liver Meeting, 11–16 November 2020.
- Damato, B.E.; Dukes, J.; Goodall, H.; Carvajal, R.D. Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma. Cancers 2019, 11, 971.
- Bertoletti, A. ImmTAV, a New Immunotherapy Targeting the Source of HBV Infection. Hepatology 2020, 72, 1514–1517.
- Lok, A.S.; Pan, C.Q.; Han, S.H.; Trinh, H.N.; Fessel, W.J.; Rodell, T.; Massetto, B.; Lin, L.; Gaggar, A.; Subramanian, G.M.; et al. Randomized phase II study of GS-4774 as a therapeutic vaccine in virally suppressed patients with chronic hepatitis B. J. Hepatol. 2016, 65, 509–516.
- Aguilar, J.C.; León, Y.; Lobaina, Y.; Freyre, F.; Fernández, G.; Sanchez, A.L.; Jerez, E.; Anillo, L.E.; Aguiar, J.A.; Cinza, Z.; et al. Five-year Follow-up of Chronic Hepatitis B Patients Immunized by Nasal Route with the Therapeutic Vaccine HeberNasvac. Euroasian J. Hepato Gastroenterol. 2018, 8, 133–139.
- Zoulim, F.; Fournier, C.; Habersetzer, F.; Sprinzl, M.; Pol, S.; Coffin, C.S.; Leroy, V.; Ma, M.; Wedemeyer, H.; Lohse, A.W.; et al. Safety and immunogenicity of the therapeutic vaccine TG1050 in chronic hepatitis B patients: A phase 1b placebo-controlled trial. Hum. Vaccines Immunother. 2020, 16, 388–399.
- Lee, H.W.; Park, J.Y.; Hong, T.; Park, M.S.; Ahn, S.H. A prospective, openlabel, dose-escalation, single-center, phase 1 study for GC1102, a recombinant human immunoglobulin for chronic hepatitis B patients. In Hepatology; WILEY: Hoboken, NJ, USA, 2018; Volume 68, pp. 268A–269A.
- Vir Biotechnology. Initial Data from Ongoing Phase 1 Trial of VIR-3434 for Chronic Hepatitis B Virus Infection Demonstrates Significant and Rapid Reduction in Hepatitis B Surface Antigen; Vir Biotechnology Inc., 2021. Available online: (accessed on 17 June 2021).
- Protzer-Knolle, U.; Naumann, U.; Bartenschlager, R.; Berg, T.; Hopf, U.; Meyer zum Buschenfelde, K.H.; Neuhaus, P.; Gerken, G. Hepatitis B virus with antigenically altered hepatitis B surface antigen is selected by high-dose hepatitis B immune globulin after liver transplantation. Hepatology 1998, 27, 254–263.
- Cooreman, M.P.; Leroux-Roels, G.; Paulij, W.P. Vaccine- and hepatitis B immune globulin-induced escape mutations of hepatitis B virus surface antigen. J. Biomed. Sci. 2001, 8, 237–247.
- de Man, R.A.; Bartholomeusz, A.I.; Niesters, H.G.; Zondervan, P.E.; Locarnini, S.A. The sequential occurrence of viral mutations in a liver transplant recipient re-infected with hepatitis B: Hepatitis B immune globulin escape, famciclovir non-response, followed by lamivudine resistance resulting in graft loss. J. Hepatol. 1998, 29, 669–675.
- Mina, T.; Amini Bavil Olyaee, S.; Tacke, F.; Maes, P.; Van Ranst, M.; Pourkarim, M.R. Genomic Diversity of Hepatitis B Virus Infection Associated With Fulminant Hepatitis B Development. Hepat. Mon. 2015, 15, e29477.
- Kucinskaite-Kodze, I.; Pleckaityte, M.; Bremer, C.M.; Seiz, P.L.; Zilnyte, M.; Bulavaite, A.; Mickiene, G.; Zvirblis, G.; Sasnauskas, K.; Glebe, D.; et al. New broadly reactive neutralizing antibodies against hepatitis B virus surface antigen. Virus Res. 2016, 211, 209–221.
- Yuen, R.; Chen, C.; Liu, C.; Jeng, R.; Elkhashab, M.; Coffin, C.; Kim, W.; Greenbloom, S.; Ramji, A.; Lim, Y.; et al. Ascending dose cohort study of inarigivir—A novel RIG I agonist in chronic HBV patients: Final results of the ACHIEVE trial. Proc. J. Hepatol. 2019, 70, e47–e48.

