Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Roman A. Blaheta | + 2043 word(s) | 2043 | 2021-06-13 06:23:27 | | | |
| 2 | Vicky Zhou | Meta information modification | 2043 | 2021-07-02 12:42:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Blaheta, R.A. Sulforaphane. Encyclopedia. Available online: https://encyclopedia.pub/entry/11611 (accessed on 10 August 2026).
Blaheta RA. Sulforaphane. Encyclopedia. Available at: https://encyclopedia.pub/entry/11611. Accessed August 10, 2026.
Blaheta, Roman A.. "Sulforaphane" Encyclopedia, https://encyclopedia.pub/entry/11611 (accessed August 10, 2026).
Blaheta, R.A. (2021, July 02). Sulforaphane. In Encyclopedia. https://encyclopedia.pub/entry/11611
Blaheta, Roman A.. "Sulforaphane." Encyclopedia. Web. 02 July, 2021.
Copy Citation
Sulforaphane (SFN) is a natural glucosinolate found in cruciferous vegetables that acts as a chemopreventive agent, but its mechanism of action is not clear. Due to antioxidative mechanisms being thought central in preventing cancer progression, SFN could play a role in oxidative processes. Since redox imbalance with increased levels of reactive oxygen species (ROS) is involved in the initiation and progression of bladder cancer, this mechanism might be involved when chemoresistance occurs.
bladder cancer
sulforaphane
ROS
oxidative stress
Keap1-Nrf2
1. Introduction
The phytochemical drug, sulforaphane (SFN), can be used to treat bladder cancer. The precursor of SFN, glucoraphanin, is highly enriched in cruciferous vegetables from the Brassicaceae family, including broccoli, cauliflower and cabbage. The mean content of glucoraphanin per gram uncooked broccoli has been calculated at 0.38 μmol, and levels of glucoraphanin evaluated in over 75 different genotypes of field-grown hybrid broccoli average between 0.88 μmol and 1.10 μmol glucoraphanin per gram fresh weight [1]. Importantly, glucoraphanin itself is not bioactive. Rather, enzymatic hydrolysis by myrosinase, present in the plant tissue or in the mammalian microbiome, is necessary to form the active component, SFN.
Ten years ago, a redox regulating function of SFN was identified [2][3]. Disrupting the redox balance by increasing free radicals, predominantly reactive oxygen species (ROS), has been shown to be closely associated with carcinogenesis and metastatic progression [4]. The degree and direction of disruption may be crucial since a moderate increase of ROS has been associated with enhanced cancer progression, while a strong and massive increase of ROS may act as a cancer suppressor by inducing apoptosis [4]. Based on in vitro, in vivo and patient studies, SFN has been shown to exert both chemopreventive and tumor-suppressive properties through multifaceted mechanisms [5].
2. SFN in Bladder Cancer
2.1. Preclinical and Clinical Studies
SFN’s role as a natural HDAC-inhibitor is highly relevant, since 90% of all cancers can be attributed to epigenetic modification [6][7]. Several US Food and Drug Administration (FDA) approved HDAC-inhibitors are currently in clinical use. Unfortunately, drugs such as Vorinostat, Belinostat, Panobinostat and Romidepsin are all associated with adverse reactions and resistance development [8]. These disadvantages could possibly be avoided by adding SFN to therapy [9]. Since SFN can easily be extracted from cruciferous vegetables and produced in large quantities, it could serve as a convenient and economic strategy for bladder cancer chemoprevention.
Cell proliferation inhibition, cell-cycle arrest, apoptosis induction, invasion and metastasis blockade all take place after bladder cancer cells are exposed to SFN [10]. SFN exerts stronger anti-proliferative effects on bladder cancer cell lines under hypoxia, compared to normoxic conditions [11]. This is important since hypoxia facilitates cancer progression, suggesting that SFN may be highly efficient in high grade, rapidly growing tumors where ROS is on the increase.
2.2. SFN’s Influence on ROS
Evidence shows that SFN upregulates the ROS level in T24 bladder cancer cells to induce apoptosis [12]. SFN treatment has been associated with loss of the mitochondrial membrane potential, with cytochrome c release and alteration of the Bcl-2/Bax ratio. In addition, SFN increases the activity of caspase-9 and -3, but not of caspase-8, and mediates the cleavage of poly ADP-ribose polymerase (PARP). These findings have been interpreted such that SFN triggered ROS generation modifies the intrinsic apoptotic pathway [12]. Other investigators have shown that SFN-induced apoptosis of 5637 cells via a ROS-dependent pathway is linked to both caspase-8 and -9 activation, indicating an influence of both intrinsic and extrinsic apoptotic pathways [13]. Therefore, it seems that ROS exerts its effects on apoptosis via different mechanisms, depending on the cell line. This means that depending on the cancer type, SFN may initiate apoptotic events in bladder cancer patients over different mechanisms and to a differing extent. It would be of interest to explore whether high and low responders to SFN can be differentiated.
2.3. SFN Acts on Nrf2
SFN potently inhibits carcinogenesis via activation of the Nrf2 pathway [14]. The daily administration of an aqueous extract of broccoli sprouts to rats (equivalent to isothiocyanate doses of 40 μmol/kg and 160 μmol/kg body weight) inhibited N-butyl-N-(4-hydroxybutyl) nitrosamine induced bladder cancer development and was associated with a significant induction of glutathione S-transferase and NAD(P)H:quinone oxidoreductase 1 [15]. Induction of these enzymes was largely mediated by Nrf2 [16]. Notably, Nrf2 activation by SFN in the bladder occurred primarily in the epithelium, which is the principal site of bladder cancer development. Since Nrf2 is critical to stimulating a variety of cytoprotective genes and is closely involved in inhibiting DNA damage, activating Nrf2 by SFN might be a key strategy to prevent bladder cancer initiation [17]. Still, the relevance of the Nrf2 pathway for bladder cancer progression is not completely understood. In fact, Nrf2 induction has also been considered a secondary process, following an increased ROS level and endoplasmic reticulum stress evoked by SFN [18]. In this case, Nrf2 could exert a prosurvival role by hindering ROS-induced apoptosis [19], and the overexpression of Nrf2 target genes could support cell proliferation by increasing ribonucleotide synthesis, serine biosynthesis and autophagy [20]. Recently, a hormetic action was found in an angiogenesis assay where 2.5 µM SFN promoted endothelial tube formation but inhibited it at 10–20 µM [21]. Whether the dose-dependency seen with SFN contributes to the role of Nrf2 as an oncoprotein or a tumor suppressor remains open.
2.4. SFN and MAPK Signaling
Although the influence of SFN on the MAPK pathway has been documented, respective experiments on bladder cancer cells are sparse. SFN upregulates the expression of two Nrf2-dependent enzymes, glutathione transferase (GSTA1-1) and thioredoxin reductase (TR-1), and downregulates COX-2 in T24 cells, which is closely associated with p38 MAPK activity [22]. Abbaoui and coworkers observed apoptosis and tumor weight reduction in murine UMUC3 xenografts exposed to SFN. The antitumor effect of SFN was associated with downregulation of both the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor 2 (HER2/neu) [23]. This is remarkable, since inhibition of either EGFR or HER2 signaling has been shown to correlate with enhanced p38 MAPK phosphorylation [24]. Gemcitabine or cisplatin treatment in human bladder cancer models has been shown to cause a dose-dependent release of ROS and activate the p38 MAPK-signaling pathway [25]. The similarity between gemcitabine/cisplatin and SFN triggered pathway alterations in bladder cancer may open new therapeutic strategies, including a combined treatment regimen to cause additive effects. SFN may also serve as an alternative drug candidate, once gemcitabine/cisplatin resistance has occurred.
2.5. SFN and NF-κB Signaling
NF-κB signaling correlates with aggressive bladder cancer behavior and poor clinical outcome [26]. Therefore, NF-κB inhibitors have been proposed as efficacious targeted therapies [27]. Concomitant NF-κB inhibition has been observed in BIU87 bladder cells, as SFN inhibits cell proliferation, arrests the cell cycle at the G2/M phase and induces apoptosis [28]. Although the underlying mode of action has not been explored in detail, the authors suggest that the insulin-like growth factor-binding protein-3 (IGFBP-3) is critically involved in suppressing NF-κB, either by blocking IGF1 signaling, by acting on cell-cycle-regulating proteins or by interfering with the MAPK-signaling pathway. SFN has been shown to downregulate COX-2 expression in T24 bladder cancer cells at both the transcriptional and translational level. This may be due to the nuclear translocation of NF-κB and reduced binding to the COX-2 promotor, initiated by upregulation of MAPK [29]. Further publication concerning SFN’s influence on the NF-κB pathway in bladder cancer is not available, so that the question of whether NF-κB inhibition is responsible for SFN’s chemopreventive and antitumor properties remains unanswered.
2.6. SFN and Akt/mTOR Modulation
The Akt-mTOR-pathway serves as a central regulator of cell growth and proliferation. In three bladder cancer cell lines (RT112, UMUC3 and TCCSUP), SFN treatment significantly suppressed the amount of phosphorylated Akt and phosphorylation of the mTOR subunit Rictor [30]. Reduction of Akt and mTOR phosphorylation, along with diminished p70S6k downstream signaling under SFN, has also been observed in HTB-9 cells [31], pointing to a common mechanism of SFN action. The relevance of SFN as a cell-cycle inhibitor has furthermore been proven in terms of diminished expression of the cell-cycle-regulating proteins of the cyclin and CDK family. Accordingly, the CDK inhibitors, p19 and p21, are elevated under SFN [30][31]. The suppressive effect of SFN on Akt-mTOR signaling has also been seen with long-term treatment, in contrast to resistance induction evoked under chronic use of the established mTOR-inhibitors, everolimus and temsirolimus [30][32]. Further investigation into adding SFN to everolimus/temsirolimus treatment for reversion or prevention of drug resistance might, therefore, be warranted. Although the Akt/mTOR pathway is connected to ROS dependent signaling, the concerted action of SFN on Akt/mTOR–ROS has not been proven. The natural compound luteolin with strong antioxidative properties has been shown to inhibit cell survival and induce G2/M cell-cycle arrest of T24 cells. This was coupled to p21 upregulation and p70S6k downregulation [33]. A similar mode of action may hold true for SFN as well. In fact, induction of apoptosis and senescence of esophageal squamous cell carcinoma cells by SFN was triggered by a ROS-mediated mTOR inactivation [34].
3. Conclusions
The natural HDAC-inhibitor, SFN, acts in a multifaceted fashion on bladder cancer, leading to cell growth arrest, proliferation blockade, apoptosis induction, along with suppression of tumor cell motility and invasion. SFN’s inhibitory activity is not restricted to bladder cancer but is apparent in other tumor types as well. Apoptosis induction by SFN via ROS is seen in hepatocellular carcinoma [35], lung cancer [36] and breast cancer cells [37]. How SFN specifically targets bladder cancer remains to be clarified. Several molecular pathways associated with bladder cancer could serve as potential targets. These include CD44-related signaling [38], Notch and MAPK signaling [39], Akt/mTOR signaling [40] or JAK/STAT and NF-κB/Snail signaling [41]. SFN influences all of these signaling pathways, making it an interesting candidate for supportive tumor therapy. Notably, negative side-effects and resistance-induction, as encountered with established drugs, are not evoked by SFN, which could further strengthen its clinical usefulness. Still, the relevance of ROS and ROS-related pathways for bladder cancer progression is not fully elucidated and both tumor-promoting and tumor-suppressing activities have been documented.
Based on current knowledge, mild elevation of ROS activates pro-tumorigenic survival and tumor growth, whereas excessive concentration of ROS leads to the induction of cell death. Thus, opposing strategies must be critically evaluated. This includes either therapeutic downregulation of ROS to prevent oncogenic signaling or upregulation of ROS above a sensitive threshold to cause oxidative damage (Figure 1).
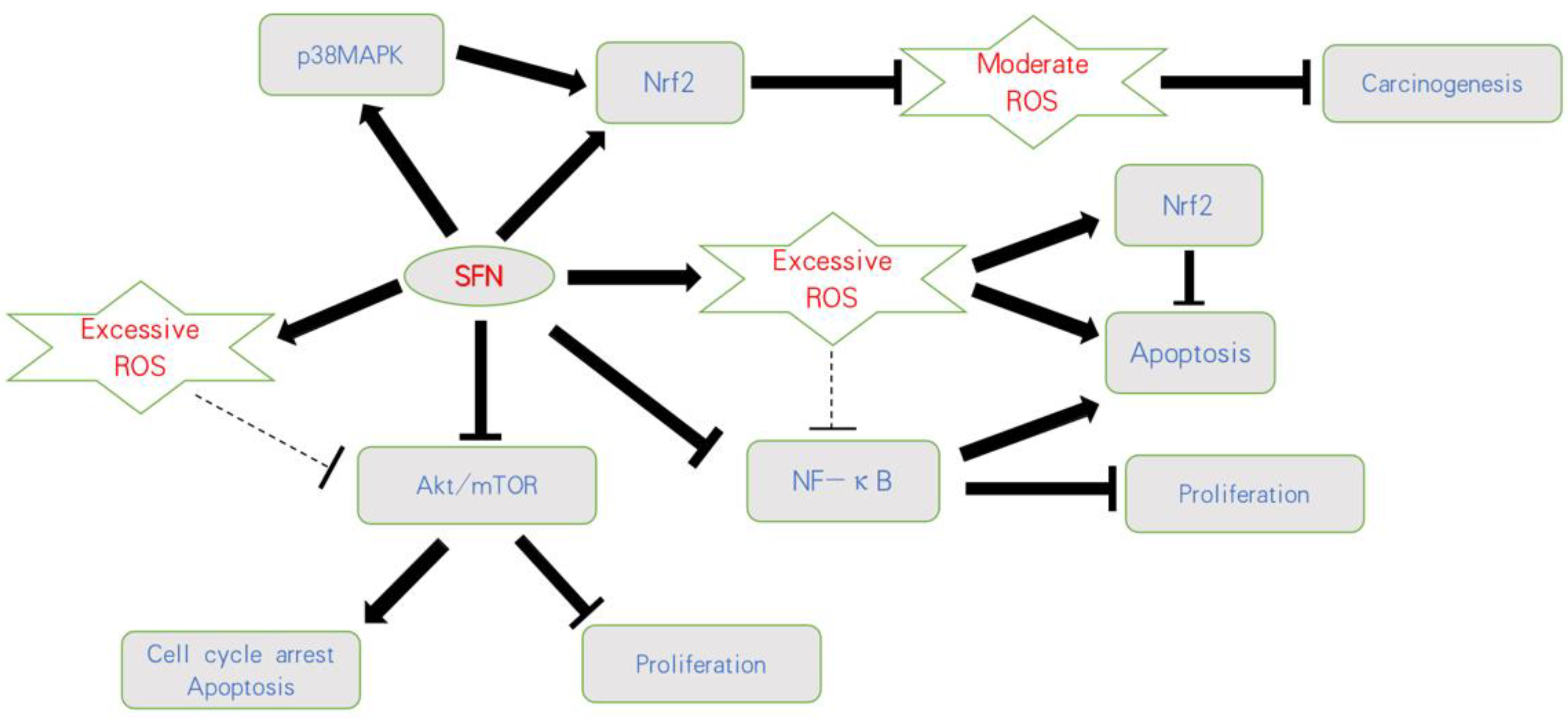 Figure 1. Influence of SFN on ROS-related pathways on bladder cancer. SFN blocks carcinogenesis by activating Nrf2 or the p38MAPK/Nrf2 axis and counteracting a moderate ROS-increase. Based on an initially excessive ROS level, SFN further increases ROS, resulting in apoptosis and proliferative inhibition. Nrf2 is thus considered a secondary product, followed by a ROS-increase involving anti-apoptotic properties. SFN also acts on the Akt/mTOR and NF-κB pathways, whereby the relevance of ROS as a trigger factor has not finally been validated;
Figure 1. Influence of SFN on ROS-related pathways on bladder cancer. SFN blocks carcinogenesis by activating Nrf2 or the p38MAPK/Nrf2 axis and counteracting a moderate ROS-increase. Based on an initially excessive ROS level, SFN further increases ROS, resulting in apoptosis and proliferative inhibition. Nrf2 is thus considered a secondary product, followed by a ROS-increase involving anti-apoptotic properties. SFN also acts on the Akt/mTOR and NF-κB pathways, whereby the relevance of ROS as a trigger factor has not finally been validated;  indicates activation;
indicates activation;  indicates inhibition;
indicates inhibition;  indicates not clear.
indicates not clear.Ongoing studies are required to precisely define the role of ROS on tumorigenesis and cancer progression. Accordingly, the consequences of SFN–ROS communication in regard to tumor cell behavior should be explored in more detail. In particular, SFN’s mode of action in tumor cells with a moderate versus substantial ROS level should be evaluated. Since the response of tumor cells to radiotherapy or chemotherapy is promoted by increased ROS production, ROS inhibition may at least be partially responsible for therapeutic resistance. In this context, treatment targeting the antioxidative stress system is an important research direction to counteract radioresistance and chemoresistance. Intriguingly, SFN has overcome cisplatin-based resistance via ROS-modulation. This is highly relevant in regard to second line treatment options. Further investigation is essential to determine the degree to which ROS contributes to the development of resistance processes triggered by undesired feedback loops and to what degree SFN counteracts tumor cell re-activation in the course of chemotherapy. Finally, SFN’s considerable antitumor potential has been documented in vitro and in vivo but not in tumor patients. Limited bioavailability of SFN remains a hurdle, necessitating further investigation into increasing bioavailability. Genetically altered plants with significantly higher amounts of glucoraphanin have been developed, which might overcome this problem. Nano-encapsulation and the synthesis of potent SFN analogues may also increase the bioavailability of SFN. Therefore, many aspects regarding SFN application remain to be investigated before a final conclusion can be drawn in respect to its use as an anticancer compound.
References
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or Sulforaphane: Is It the Source or Dose That Matters? Molecules 2019, 24, 3593.
- Wang, Y.; Mandal, A.K.; Son, Y.O.; Pratheeshkumar, P.; Wise, J.T.F.; Wang, L.; Zhang, Z.; Shi, X.; Chen, Z. Roles of ROS, Nrf2, and autophagy in cadmium-carcinogenesis and its prevention by sulforaphane. Toxicol. Appl. Pharmacol. 2018, 353, 23–30.
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735.
- Juengel, E.; Erb, H.H.H.; Haferkamp, A.; Rutz, J.; Chun, F.K.; Blaheta, R.A. Relevance of the natural HDAC inhibitor sulforaphane as a chemopreventive agent in urologic tumors. Cancer Lett. 2018, 435, 121–126.
- Paul, B.; Li, Y.; Tollefsbol, T.O. The Effects of Combinatorial Genistein and Sulforaphane in Breast Tumor Inhibition: Role in Epigenetic Regulation. Int. J. Mol. Sci. 2018, 19, 1754.
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. 2009, 15, 425–430.
- Ranganna, K.; Selvam, C.; Shivachar, A.; Yousefipour, Z. Histone Deacetylase Inhibitors as Multitarget-Directed Epi-Drugs in Blocking PI3K Oncogenic Signaling: A Polypharmacology Approach. Int. J. Mol. Sci. 2020, 21, 8198.
- Rutz, J.; Juengel, E.; Euler, S.; Maxeiner, S.; Justin, S.; Roos, F.; Chun, F.K.; Blaheta, R.A. Chronic Sulforaphane Application Does Not Induce Resistance in Renal Cell Carcinoma Cells. Anticancer Res. 2018, 38, 6201–6207.
- Mastuo, T.; Miyata, Y.; Yuno, T.; Mukae, Y.; Otsubo, A.; Mitsunari, K.; Ohba, K.; Sakai, H. Molecular Mechanisms of the Anti-Cancer Effects of Isothiocyanates from Cruciferous Vegetables in Bladder Cancer. Molecules 2020, 25, 575.
- Xia, Y.; Kang, T.W.; Jung, Y.D.; Zhang, C.; Lian, S. Sulforaphane Inhibits Nonmuscle Invasive Bladder Cancer Cells Proliferation through Suppression of HIF-1α-Mediated Glycolysis in Hypoxia. J. Agric. Food Chem. 2019, 67, 7844–7854.
- Shan, Y.; Zhang, L.; Bao, Y.; Li, B.; He, C.; Gao, M.; Feng, X.; Xu, W.; Zhang, X.; Wang, S. Epithelial-mesenchymal transition, a novel target of sulforaphane via COX-2/MMP2, 9/Snail, ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J. Nutr. Biochem. 2013, 24, 1062–1069.
- Park, H.S.; Han, M.H.; Kim, G.Y.; Moon, S.K.; Kim, W.J.; Hwang, H.J.; Park, K.Y.; Choi, Y.H. Sulforaphane induces reactive oxygen species-mediated mitotic arrest and subsequent apoptosis in human bladder cancer 5637 cells. Food Chem. Toxicol. 2014, 64, 157–165.
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and Done? Targeting the NRF2 Pathway with Sulforaphane. Trends Food Sci. Technol. 2017, 69 Pt B, 257–269.
- Munday, R.; Mhawech-Fauceglia, P.; Munday, C.M.; Paonessa, J.D.; Tang, L.; Munday, J.S.; Lister, C.; Wilson, P.; Fahey, J.W.; Davis, W.; et al. Inhibition of urinary bladder carcinogenesis by broccoli sprouts. Cancer Res. 2008, 68, 1593–1600.
- Zhang, Y.; Munday, R.; Jobson, H.E.; Munday, C.M.; Lister, C.; Wilson, P.; Fahey, J.W.; Mhawech-Fauceglia, P. Induction of GST and NQO1 in cultured bladder cells and in the urinary bladders of rats by an extract of broccoli (Brassica oleracea italica) sprouts. J. Agric. Food Chem. 2006, 54, 9370–9376.
- Ding, Y.; Paonessa, J.D.; Randall, K.L.; Argoti, D.; Chen, L.; Vouros, P.; Zhang, Y. Sulforaphane inhibits 4-aminobiphenyl-induced DNA damage in bladder cells and tissues. Carcinogenesis 2010, 31, 1999–2003.
- Jo, G.H.; Kim, G.Y.; Kim, W.J.; Park, K.Y.; Choi, Y.H. Sulforaphane induces apoptosis in T24 human urinary bladder cancer cells through a reactive oxygen species-mediated mitochondrial pathway: The involvement of endoplasmic reticulum stress and the Nrf2 signaling pathway. Int. J. Oncol. 2014, 45, 1497–1506.
- Jin, C.Y.; Molagoda, I.M.N.; Karunarathne, W.; Kang, S.H.; Park, C.; Kim, G.Y.; Choi, Y.H. TRAIL attenuates sulforaphane-mediated Nrf2 and sustains ROS generation, leading to apoptosis of TRAIL-resistant human bladder cancer cells. Toxicol. Appl. Pharmacol. 2018, 352, 132–141.
- Robertson, H.; Dinkova-Kostova, A.T.; Hayes, J.D. NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis. Cancers 2020, 12, 3609.
- Bao, Y.; Wang, W.; Zhou, Z.; Sun, C. Benefits and risks of the hormetic effects of dietary isothiocyanates on cancer prevention. PLoS ONE 2014, 9, e114764.
- Shan, Y.; Wang, X.; Wang, W.; He, C.; Bao, Y. p38 MAPK plays a distinct role in sulforaphane-induced up-regulation of ARE-dependent enzymes and down-regulation of COX-2 in human bladder cancer cells. Oncol. Rep. 2010, 23, 1133–1138.
- Abbaoui, B.; Riedl, K.M.; Ralston, R.A.; Thomas-Ahner, J.M.; Schwartz, S.J.; Clinton, S.K.; Mortazavi, A. Inhibition of bladder cancer by broccoli isothiocyanates sulforaphane and erucin: Characterization, metabolism, and interconversion. Mol. Nutr. Food Res. 2012, 56, 1675–1687.
- Mora Vidal, R.; Regufe da Mota, S.; Hayden, A.; Markham, H.; Douglas, J.; Packham, G.; Crabb, S.J. Epidermal Growth Factor Receptor Family Inhibition Identifies P38 Mitogen-activated Protein Kinase as a Potential Therapeutic Target in Bladder Cancer. Urology 2018, 112, e1–e225.
- Wang, X.; Chen, F.; Gou, S. Anti-tumor effects and cell motility inhibition of the DN604-gemcitabine combined treatment in human bladder cancer models. Bioorg. Med. Chem. 2021, 29, 115858.
- Kim, B.; Jang, I.; Kim, K.; Jung, M.; Lee, C.; Park, J.H.; Kim, Y.A.; Moon, K.C. Comprehensive Gene Expression Analyses of Immunohistochemically Defined Subgroups of Muscle-Invasive Urinary Bladder Urothelial Carcinoma. Int. J. Mol. Sci. 2021, 22, 628.
- Lyu, Z.J.; Wang, Y.; Huang, J.L.; Chen, M.; Wu, S.Y.; Yan, Q.; Zhang, Y.; Tang, Y.; Jiang, C.; Li, L.; et al. Recurrent ZNF83-E293V Mutation Promotes Bladder Cancer Progression through the NF-κB Pathway via Transcriptional Dysregulation of S100A8. Mol. Ther. 2021, 29, 275–290.
- Dang, Y.M.; Huang, G.; Chen, Y.R.; Dang, Z.F.; Chen, C.; Liu, F.L.; Guo, Y.F.; Xie, X.D. Sulforaphane inhibits the proliferation of the BIU87 bladder cancer cell line via IGFBP-3 elevation. Asian Pac. J. Cancer Prev. 2014, 15, 1517–1520.
- Shan, Y.; Wu, K.; Wang, W.; Wang, S.; Lin, N.; Zhao, R.; Cassidy, A.; Bao, Y. Sulforaphane down-regulates COX-2 expression by activating p38 and inhibiting NF-kappaB-DNA-binding activity in human bladder T24 cells. Int. J. Oncol. 2009, 34, 1129–1134.
- Justin, S.; Rutz, J.; Maxeiner, S.; Chun, F.K.; Juengel, E.; Blaheta, R.A. Chronic Sulforaphane Administration Inhibits Resistance to the mTOR-Inhibitor Everolimus in Bladder Cancer Cells. Int. J. Mol. Sci. 2020, 21, 4026.
- Islam, S.S.; Mokhtari, R.B.; Akbari, P.; Hatina, J.; Yeger, H.; Farhat, W.A. Simultaneous Targeting of Bladder Tumor Growth, Survival, and Epithelial-to-Mesenchymal Transition with a Novel Therapeutic Combination of Acetazolamide (AZ) and Sulforaphane (SFN). Target Oncol. 2016, 11, 209–227.
- Juengel, E.; Natsheh, I.; Najafi, R.; Rutz, J.; Tsaur, I.; Haferkamp, A.; Chun, F.K.; Blaheta, R.A. Mechanisms behind Temsirolimus Resistance Causing Reactivated Growth and Invasive Behavior of Bladder Cancer Cells In Vitro. Cancers 2019, 11, 777.
- Iida, K.; Naiki, T.; Naiki-Ito, A.; Suzuki, S.; Kato, H.; Nozaki, S.; Nagai, T.; Etani, T.; Nagayasu, Y.; Ando, R.; et al. Luteolin suppresses bladder cancer growth via regulation of mechanistic target of rapamycin pathway. Cancer Sci. 2020, 111, 1165–1179.
- Zheng, K.; Ma, J.; Wang, Y.; He, Z.; Deng, K. Sulforaphane Inhibits Autophagy and Induces Exosome-Mediated Paracrine Senescence via Regulating mTOR/TFE3. Mol. Nutr. Food Res. 2020, 64, e1901231.
- Hwangbo, H.; Kim, S.Y.; Lee, H.; Park, S.H.; Hong, S.H.; Park, C.; Kim, G.Y.; Leem, S.H.; Hyun, J.W.; Cheong, J.; et al. Auranofin Enhances Sulforaphane-Mediated Apoptosis in Hepatocellular Carcinoma Hep3B Cells through Inactivation of the PI3K/Akt Signaling Pathway. Biomol. Ther. 2020, 28, 443–455.
- Wang, T.H.; Chen, C.C.; Huang, K.Y.; Shih, Y.M.; Chen, C.Y. High levels of EGFR prevent sulforaphane-induced reactive oxygen species-mediated apoptosis in non-small-cell lung cancer cells. Phytomedicine 2019, 64, 152926.
- Jabbarzadeh Kaboli, P.; Afzalipour Khoshkbejari, M.; Mohammadi, M.; Abiri, A.; Mokhtarian, R.; Vazifemand, R.; Amanollahi, S.; Yazdi Sani, S.; Li, M.; Zhao, Y.; et al. Targets and mechanisms of sulforaphane derivatives obtained from cruciferous plants with special focus on breast cancer—Contradictory effects and future perspectives. Biomed. Pharmacother. 2020, 121, 109635.
- Sottnik, J.L.; Vanderlinden, L.A.; Joshi, M.; Chauca-Diaz, A.; Owens, C.; Hansel, D.E.; Sempeck, C.; Ghosh, D.; Theodorescu, D. Androgen Receptor Regulates CD44 Expression in Bladder Cancer. Cancer Res. 2021.
- Schulz, G.B.; Elezkurtaj, S.; Börding, T.; Schmidt, E.M.; Elmasry, M.; Stief, C.G.; Kirchner, T.; Karl, A.; Horst, D. Therapeutic and prognostic implications of NOTCH and MAPK signaling in bladder cancer. Cancer Sci. 2021, 112, 1987–1996.
- Liu, X.; Zhang, W.; Wang, H.; Lai, C.H.; Xu, K.; Hu, H. Increased expression of POLR3G predicts poor prognosis in transitional cell carcinoma. PeerJ 2020, 8, e10281.
- Taheri, M.; Shirvani-Farsani, Z.; Ghafouri-Fard, S.; Omrani, M.D. Expression profile of microRNAs in bladder cancer and their application as biomarkers. Biomed. Pharmacother. 2020, 131, 110703.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
02 Jul 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No