Novel treatment regimens are required for castration-resistant prostate cancers (CRPCs) that become unresponsive to standard treatments, such as docetaxel and enzalutamide. Histone deacetylase (HDAC) inhibitors showed promising results in hematological malignancies, but they failed in solid tumors such as prostate cancer, despite the overexpression of HDACs in CRPC. Four HDAC inhibitors, vorinostat, pracinostat, panobinostat and romidepsin, underwent phase II clinical trials for prostate cancers; however, phase III trials were not recommended due to a majority of patients exhibiting either toxicity or disease progression. In this entry, the pharmacodynamic reasons for the failure of HDAC inhibitors were assessed and placed in the context of the advancements in the understanding of CRPCs, HDACs and resistance mechanisms.
1. Introduction
There were ~1.3 million new cases of prostate cancer worldwide in 2018 and it is the second most commonly diagnosed cancer in men
[1]. Importantly, it ranks second among all cancer fatalities in males
[2]. Even though the five-year survival for localized disease is 100%, it drops down to 29.3% if the prostate cancer metastasizes to other organs
[3]. Furthermore, from 2005 to 2018 a 31% increase in the incidence of prostate cancer was observed from 974,000 to 1.3 million new cases
[1][4]. The majority of prostate cancers are initially androgen-dependent
[5]. In advanced prostate cancer, androgen deprivation therapy, either by chemical castration in the form of luteinizing hormone-releasing hormone agonists/antagonists or surgical castration has typically been used. After a median of 18–24 months of treatment, most patients progress to a metastatic castration-resistant prostate cancer (CRPC)
[6][7], which has a median survival time of 23–37 months
[2]. Therefore, there is a need for novel therapeutic strategies that exploit the molecular signature of prostate cancers.
One of these strategies involves modulating histone acetylation and deacetylation mediated by histone acetyl transferases (HATs) and histone deacetylases (HDACs), respectively. HATs and HDACs affect both, histone and non-histone proteins
[8]. Histone proteins, H2A, H2B, H3 and H4, form the core of the nucleosomes with DNA wrapped around them to form chromatin
[9].
The histone core proteins have lysine residues on their N-terminal tails
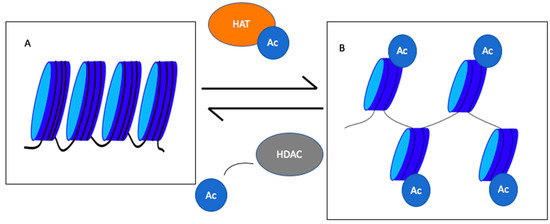
[10]. If these lysines are not acetylated, histones remain positively charged and ionically interact with the negatively-charged phosphate backbone of DNA (Figure 1). In this case, chromatin will remain condensed and the transcription machinery will not be able to access the underlying transcription site. Acetylation of lysine residues leads to chromatin decondensation and transcription initiation
[11]. Importantly, the balance in the transcription of oncogenes and tumor suppressor genes is lost in cancer
[12]. In addition to histones, non-histone proteins can be acetylated. Acetylation of transcription factors, such as p-53, NF- κB, p50 and PC4, increases their sequence-specific binding to the DNA, whereas acetylation of other factors, such as FoxO1, HMGI (Y) and p65, reduces their ability to bind DNA
[8]. Regulation of acetylation via histones and non-histone proteins can have a wide repertoire of effects, including altered gene transcription, DNA damage repair, cell division, signal transduction, protein folding, autophagy and metabolism
[13][14]. With such a wide range of actions, understanding the mechanisms of protein acetylation and deacetylation is essential to devise new therapies.
Figure 1. Roles of histone acetyl transferases (HATs) and histone deacetylase (HDACs) in chromatin condensation and decondensation. HATs add an acetyl group to histones decondensing chromatin, whereas HDACs remove acetyl groups leading to chromatin condensation. (
A) represents the condensed chromatin, and (
B) shows the decondensed chromatin. Ac = acetyl group
[11].
2. HDAC Inhibitors
As HDAC expression has been shown to be associated with poor clinical outcome, HDAC inhibitors have been explored as a potential therapeutic option. The five classes of HDAC inhibitors include hydroxamic acids, cyclic tetrapeptides, short chain carboxylic acids, benzamides and keto-derivatives
[15]. These inhibitors have a well accepted pharmacophore, consisting of a zinc-binding group, coordinating with the zinc ion in the active site, a linker that transverses the active site and a cap for interactions with the external surface
[16].
Due to the role of HDAC inhibitors in inducing cell cycle arrest, apoptosis, autophagy, heat shock protein-90 (HSP90) inhibition and reactive oxygen species generation, this class of drugs has been trialed in cancers
[17]. In 2006, the FDA approved the use of a hydroxamic acid drug, suberanilohydroxamic acid (SAHA, vorinostat), for the treatment of cutaneous T-cell lymphoma
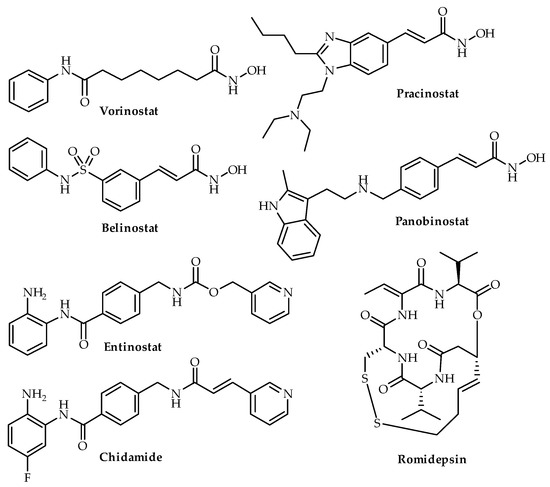
[18]. Since then, HDAC inhibitors, including belinostat, panobinostat and romidepsin, have been approved for peripheral T-cell cutaneous lymphoma and multiple myeloma (Figure 2). Hematological malignancies have shown promising responses to HDAC inhibitors
[19]. However, HDAC inhibitors have not successfully cleared clinical trials for solid tumors, despite promising collective results in biologic, preclinical and phase I and II studies
[17][20]. For example, in phase III trials for advanced hormone receptor-positive breast cancer, chidamide, a HDAC inhibitor, in combination with exemestane, increased the median progression-free survival to 7.4 months in comparison to 3.8 months with placebo and has been recommended for further testing
[21]. Other HDAC inhibitors are undergoing trials in solid tumors, including entinostat in phase III trials for locally advanced or metastatic recurrent hormone receptor-positive breast cancer (Table 1)
[22].
Figure 2. Chemical structures of four HDAC inhibitors (vorinostat, belinostat, romidepsin, pracinostat, entinostat, chidamide and panobinostat). Vorinostat, belinostat, romidepsin and panobinostat are FDA-approved.
Table 1. HDAC inhibitors undergoing clinical trials.
| HDAC Inhibitor |
Drug Combination |
Tumor Type |
Trial Phase Completed [Reference] |
| Entinostat |
Exesmestane |
Recurrent hormone receptor-positive breast cancer |
Phase II trial [23] |
| Permolizumab |
Metastatic uveal melanoma |
Phase II trial [24] |
| Chidamide |
Exesmestane |
Hormone receptor-positive, HER2-negative breast cancer |
Phase III trial [21] |
| Vorinostat |
Permolizumab |
Advanced/metastatic Non-small cell lung cancer |
Phase I trial [25] |
SAHA, romidepsin, panobinostat and pracinostat (Figure 2) are the four HDAC inhibitors that have undergone clinical trials for CRPC and are discussed below. Apart from romidepsin, SAHA, panobinostat and pracinostat are classed as hydroxamic acid derivatives. There is no clear-cut description of structure activity relationship for testing mainly hydroxamic acid derivatives in the clinical trials conducted for CRPC. Qian and colleagues (2016) recommended entinostat, a benzamide, for clinical testing in prostate cancer based on in vitro and in vivo experiments
[26]. Details around the effect of class and structure of HDAC inhibitors remain unexplored. No trends can be observed in the IC
50 values of different classes of HDAC inhibitors. Hydroxamic acid derivatives, including SAHA, panobinostat, belinostat and pracinostat, inhibit different classes with different efficacies; similarly, different compounds from the benzamide class, including chidamide, entinostat and mocetinostat exhibit different affinities for different HDACs (Table 2). Even while considering a pharmacokinetic parameter such as half-life. Chidamide, entinostat and mocetinostat had a half-life of 16–18, 51.58 and 7–11 h, whereas vorinostat, panobinostat, pracinostat and belinostat had a half-life of 0.8–3.9, 12, 5.6–8.9 and 0.9 h
[27][28][29].
Table 2. IC
50 values of HDAC inhibition
[16][30][31][32].
| Compound |
HDAC Inhibition IC50 (nM) |
| Class I |
Class IIa |
Class IIb |
Class IV |
| 1 |
2 |
3 |
8 |
4 |
5 |
7 |
9 |
6 |
10 |
11 |
| SAHA |
38 |
144 |
6 |
38 |
>30000 |
>30000 |
>300000 |
>30000 |
10 |
21 |
28 |
| Panobinostat |
3 |
3 |
4 |
248 |
23 |
NA |
18 |
6 |
3 |
ND |
ND |
| Belinostat |
41 |
125 |
30 |
216 |
115 |
NA |
67 |
128 |
82 |
ND |
ND |
| Pracinostat |
49 |
96 |
43 |
140 |
56 |
47 |
137 |
70 |
1008 |
40 |
ND |
| Chidamide |
95 |
160 |
67 |
733 |
>30000 |
>30000 |
>30000 |
>30000 |
>30000 |
78 |
432 |
| Entinostat |
262 |
306 |
499 |
2700 |
>30000 |
>30000 |
>30000 |
>30000 |
>30000 |
254 |
0.649 |
| Mocetinostat |
150 |
290 |
1660 |
>10000 |
>10000 |
>10000 |
>10000 |
ND |
ND |
ND |
590 |
| Romidepsin |
36 |
47 |
ND |
ND |
510 |
ND |
ND |
ND |
14000 |
ND |
ND |
3. Pharmacodynamic Rationale for Treatment Failure Using HDAC Inhibitors
As a single agent, HDAC inhibitors have shown poor activity in CRPC and other solid tumors
[2][33]. Previously, this has been attributed to their pharmacokinetic profile
[34]. This argument can be made in the case of SAHA, which is classified as a class IV drug in the Biopharmaceutical Classification System, due to its low water solubility (190 µg/mL) and low cell permeability (2 × 10
6 cm/s)
[29]. Among other parameters, the drug exhibits poor oral bioavailability (around 11% and 2% in the dog and rat, respectively) and a short half-life of 12 min. Similarly, panobinostat exhibits poor oral bioavailability of 6% in rats and a moderate bioavailability of 33–50% in dogs, whereas romidepsin has not been administered orally in animals due to poor solubility
[35]. However, pracinostat has been classified as a BioPharmaceutical Classification System class I compound, as it has a higher water solubility, higher oral bioavailability (34% and 65% in mice and dogs, respectively) and longer half-life of 4.1 h in dogs
[29][36][37]. It is worth noting, however, that while some patients exhibited disease progression, others have shown disease stabilization and PSA declines >50%
[20][38][39][40]. Given the conflicting results, the pharmacodynamic profile of HDAC inhibitors is clearly favorable in some patients.
Recently, new insights have been shed on prostate cancer, bringing the centrality of AR, effects on different classes of HDACs by non-specific HDAC inhibitors and evolution of resistance mechanisms in response to HDAC inhibitors into question.
3.1. AR-Negative Prostate Cancers
AR is thought to play a central role in the progression of prostate cancers
[41]. Most studies show HDAC-mediated inhibition of HSP-90 acetylation as one of the main mechanisms by which AR can be regulated
[38][40]. However, studies report different percentages of AR expression in CRPC. In a study conducted by Leav and colleagues (2001), AR immunostaining was present in more than 95% of metastatic cells irrespective of tumor grade
[42]. Pronounced AR staining in the nucleus was also seen in normal prostate secretory and stromal cells
[42]. In contrast, a study conducted by Shah and colleagues (2004) indicated that AR expression was variable among patients who died from metastasized CRPC. In this study, AR staining was less than 10% in 41.6% of patients, which suggests that there are alternative AR bypass mechanisms
[43]. However, no comparisons were made between different regions of the organs to show differences between adjacent tumor sites
[43].
These findings are also supported by another study which showed that serum PSA declines of >50% were observed in 56% of 140 patients with metastatic CRPC following treatment with the AR antagonist enzalutamide
[44]. The baseline CTC population decreased by 75% of chemotherapy-naïve and 37% of post-chemotherapy patients
[43]. Interestingly, tumors classified from the pre-enzalutamide era (1997–2011) differed in molecular signatures from tumor tissues obtained after the approval of enzalutamide
[41]. Data pooled from immunohistochemistry and RNA sequencing of 56 patients showed that 88.4% of patients expressed an AR+ prostate cancer, whereas 6.3% had a neuroendocrine (NE) variant and 6.3% had an AR-/NE- variant from the period of 1998-Compared with data from 2012 to 2016, the figure dropped to 63.3% of patients expressing AR, whereas the AR-/NE+ variant rose to 13.3% and the AR-/NE- variant rose to 23.3%
[41]. Although enzalutamide-resistant cell lines have not been studied, knock-down models, including the knockdown of the AR in LnCAP cells, were investigated with the aim of understanding the AR bypass mechanisms. The FGF8/MAPK pathway was implicated as a major AR bypass mechanism in cell lines and a patient-derived xenograft model, however, studies in patients pertaining to this pathway have not been conducted
[41].
Interestingly, there has been no correlation between AR and PSA, which may just be a trend seen in late stage prostate cancer
[41]. However, it has been widely reported that AR is responsible for regulating PSA expression via indirect and direct means
[41][45][46][47]. Given the shifting paradigm in the understanding of prostate cancer, the percentage expression of AR requires further investigation. To date, clinical trials have not studied AR expression. It will also be interesting to understand the correlation between AR expression and HDAC inhibition, given that HDAC plays a role in HSP90 acetylation.
3.2. Other Cellular Targets of HDACs
Although HDAC inhibitors induce apoptosis and cell cycle arrest in various tumors, they also lead to upregulation of epithelial to mesenchymal (EMT) transitioning proteins and confer protective effects in cancer cells via p21 inductions
[48][49][50]. These findings can be attributed to the different functions of HDAC classes, as presented in the studies below. Noteworthily, the HDAC inhibitors, SAHA, romidepsin, panobinostat and pracinostat that underwent trials are not specific and inhibit a range of HDACs (Table 3)
[16]. Therefore, rather than inhibiting tumor growth, HDAC inhibitors may facilitate its growth.
Table 3. HDAC inhibitors examined in clinical trials to treat prostate cancer
[10].
| Name |
Structural Class |
HDACs Inhibited |
Prostate Cancer Clinical Trial Status |
| SAHA |
Hydroxamic acid |
I, IIb and IV |
II failed |
| Depsipeptide (Romidepsin) |
Cyclic peptide |
HDAC 1, 2 and 4 |
II failed |
| Pracinostat (SB939) |
Hydroxamic acid |
I, IIa and HDAC10 |
II failed |
| Panobinostat |
Hydroxamic acid |
HDAC I, 4, 6, 7 and 9 |
II failed |
Various studies support these theories. Kong and colleagues (2012) showed that HDAC inhibitors caused acetylation of histone 3 residues in the promoter regions of EMT-related factors such as vimentin, zinc finger E-box-binding homeobox 1 (ZEB1), Slug and matrix metalloproteinase (MMP2). The Western blotting analysis also revealed that the levels of HDAC1 and HDAC2 were unchanged and interacted with the promoters of vimentin, Slug and ZEB1 following treatment with trichostatin A (TSA) and SAHA. These findings suggested that the activity of HDACs was repressed, but the amount of HDACs were unchanged
[48]. Using immunofluorescence and reverse transcription polymerase chain reaction, an accumulation of vimentin, ZEB1 and F-actin was observed in response to treatments with SAHA and TSA at concentrations of 5 µM and 400 nM, respectively, in PC-3 cells
[48]. Following treatment with SAHA or TSA, PC3, Du145 and ARCaPE cells showed an irregular fibroblastoid morphology, reminiscent of EMT. As PC-3 cells are AR- cells, AR+ LnCAP cells were also treated with SAHA (2.5 and 5 µM) and TSA (200 and 400 nM). Increases in the mRNA expressions of ZEB1, Slug, vimentin and N-cadherin were seen, along with increasing protein expression of vimentin and fibronectin 24 h after treatment
[48]. Nasopharyngeal, colon, liver and lung cancers have shown similar responses to sodium butyrate, valproic acid (VPA) and SAHA at similar concentrations via upregulations in Snail and Slug
[51]. Therefore, active HDAC1 and 2 are capable of inhibiting EMT, and HDAC inhibitors can induce EMT.
Of the few studies examining changes in angiogenesis elicited by HDAC inhibitors, most of the studies have focused on modulating the expression levels of hypoxia inducible factor-1α (HIF-1α), vascular endothelial growth factor receptor (VEGFR)-2 and vascular endothelial growth factor (VEGF)-A in cancer cell lines
[52][53]. Mixed results have been reported in response to different drugs. Specifically, SAHA (2.5–10 µM) reduced VEGFA protein expression in lung cancer cell lines (PC9, HCC827, H1975 and NCI-H460)
[52][53]. Interestingly, SAHA (5 mmol/L) and VPA (1 mmol/L) caused sprouting of endothelial cell spheroids from HUVECs via activation of β-catenin
[54]. VPA was used at this concentration as it equates to patient plasma levels
[54]. A further rationale for these studies was the premise that HDAC5 and HDAC7 induce endothelial cell migration
[54]. In another study, SAHA (5 µM) and TSA (400 nM) were able to stop the invasion of HUVECs into type I collagen gel
[55]. TSA also inhibited the sprouting of capillaries from ex vivo rat aortic rings and VEGF-induced expression of VEGFR1, VEGFR2 and neuropilin-1
[55]. Furthermore, the VEGF competitive inhibitor, semaphorin, was upregulated by SAHA and TSA in HUVECs
[55]. Interestingly, the differential effects of HDAC inhibitors have been linked to their concentrations. Aurora and colleagues (2010) showed that HDAC inhibitors at low concentrations synergized with the competitive inhibitor of angiogenesis, pigment epithelium-derived factor (PEDF), whereas at higher concentrations HDAC inhibitors antagonized PEDF
[56].
Various class I and II HDACs exert anti-angiogenic effects
[57]. Kruppel-like factor 4 (KLF4) expression in breast cancer modulates VEGF activity via HDAC2 and HDAC3. Ray and colleagues (2013) showed that an ectopic expression of individual KLF4, HDAC2 and HDAC3 decreased VEGF-CAT reporter activity in MCF-10A cells
[58]. Chromatin immunoprecipitation (ChIP) analysis also revealed that KLF4, HDAC2 and HDAC3 interacted with the VEGF promoter
[58]. In another setting, HDAC5 interacted with the promoter of FGF2 and SLIT2, factors that are critical for endothelial cell growth, as determined by microarray analysis and ChIP assays
[59]. Accordingly, knockdown of HDAC5 by siRNA led to HUVEC sprouting
[56]. Furthermore, silencing of HDAC7, a class II HDAC, led to an up-regulation of the pro-angiogenic factor, platelet derived growth factor-B (PDGF-B) in HUVEC cells
[60].
The studies discussed above demonstrate that HDACs can in fact exert anti-tumorigenic effects, as seen in studies discussed above. Table 1 sums up the different functions of HDAC subtypes to further reinforce that HDACs can exert both anti-tumorigenic and pro-tumorigenic effects.
 +1 credit
+1 credit