Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Massimo Conese | + 4156 word(s) | 4156 | 2021-04-23 05:42:00 | | | |
| 2 | Karina Chen | Meta information modification | 4156 | 2021-07-05 04:09:42 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Conese, M. Lung Disease in Cystic Fibrosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/11526 (accessed on 09 August 2026).
Conese M. Lung Disease in Cystic Fibrosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/11526. Accessed August 09, 2026.
Conese, Massimo. "Lung Disease in Cystic Fibrosis" Encyclopedia, https://encyclopedia.pub/entry/11526 (accessed August 09, 2026).
Conese, M. (2021, June 30). Lung Disease in Cystic Fibrosis. In Encyclopedia. https://encyclopedia.pub/entry/11526
Conese, Massimo. "Lung Disease in Cystic Fibrosis." Encyclopedia. Web. 30 June, 2021.
Copy Citation
Cystic fibrosis (CF) is an autosomal recessive, life-threatening condition affecting many organs and tissues, the lung disease being the chief cause of morbidity and mortality. Mutations affecting the CF Transmembrane Conductance Regulator (CFTR) gene determine the expression of a dysfunctional protein that, in turn, triggers a pathophysiological cascade, leading to airway epithelium injury and remodeling.
cystic fibrosis
CFTR
airway epithelium
wound healing
EGF/EGFR
epithelial-mesenchymal transition
curcumin
CFTR modulators
mesenchymal stem cells
1. Introduction
Cystic fibrosis (CF) is the most common autosomal recessive disorder in the Caucasian population, affecting more than 80,000 people around the world. CF affects many mucosal organs lined by an epithelium, including the lung and gastrointestinal tract, as well as many glands with secretory/adsorptive functions. The lung disease is, however, the major determinant of morbidity and mortality of CF individuals [1]. More than 2000 variants of the CFTR gene have been described, with a genotype-phenotype correlation for only 322 mutations (https://www.cftr2.org/, accessed on 4 March 2021) organized into six classes depending on the fate and function of the CFTR protein [2]. According to a newer classification [3], which includes a previous proposal of De Boeck and Amaral [4], CFTR mutations are still categorized into six classes, with the only exception that class I now comprises those of class 1A (no mRNA transcription) and class 1B (stop-codon mutations), both having the same outcome, i.e., absence of the CFTR protein (in case of class IB due to degradation of truncated mRNA by nonsense-mediated decay). Class II (to which the most common mutation—F508del—belongs) include mutants that do not overpass the endoplasmic reticulum quality control and are degraded in the proteasome. Class III and class IV mutations are those impairing gating or alter conductance of chloride and bicarbonate ions, respectively, of a CFTR channel correctly transported to the apical membrane. Class V mutations lead to a reduction in the CFTR protein levels due to alternative splicing. Class VI comprises those mutations that destabilize the CFTR protein at the apical membrane. Classes I, II, and III are associated with a more severe phenotype, whereas classes IV, V, and VI with milder phenotypes.
Since the discovery of the CFTR gene in 1989, many studies have elucidated the pathophysiological cascade occurring in the bronchi/bronchioli. The CFTR protein is a transmembrane glycoprotein which is comprised of two specular halves (each composed of six membrane-spanning domains and a nucleotide binding domain) connected by a regulatory domain, which makes it unique among the other members of the superfamily of ATP-binding cassette proteins. Functions classically ascribed to CFTR are linked to its activity as a channel of chloride and bicarbonate ions, involved in the proper hydration of airway surface liquid (ASL) [5]. Low or null CFTR protein expressed at the plasma membrane or its dysfunction [4] results in airway surface dehydration and ASL volume depletion, whose pathogenesis is attributable to abnormal ion and fluid homeostasis at the apical side of airway epithelial cells [6]. CFTR is involved also in sodium ions concentration in the ASL by a tonic inhibitory effect on the epithelial sodium channel (ENaC) [7][8], thereby in CF ENaC is hyper-activated and contributes to the liquid hypersorption from the airways [9]. Moreover, loss or dysfunctional CFTR is responsible for defective bicarbonate secretion, resulting in a reduced antimicrobial activity of ASL [10], as well as alterations in rheological properties of mucus which becomes dense with increased viscosity [11]. Altogether, these early events ultimately lead to reduced mucociliary clearance (MCC), mucus accumulation, airway plugging, bacterial colonisation, neutrophil inflammation, progressive tissue damage and decline in lung function in CF airways. A wealth of inflammatory and remodeling mediators has been found at altered levels in CF airways, i.e., in samples of sputum, bronchoalveolar lavage (BAL) fluid and exhaled breath condensate, as well as in the blood [12]. These mediators reflect the recruitment and activation of neutrophils (for example, HMGB-1, IL-1β, TNF-α, IL-8, GM-CSF, IL-17, elastase, myeloperoxidase), activation of adaptive immunity arms (IFN-γ, IL-17, IL-33), and events linked to angiogenesis and fibrosis (VEGF, TGF-β).
Besides its role as a chloride/bicarbonate channel, CFTR is known to be involved in many cellular and tissue processes, such as fetal development [13], epithelial differentiation/polarization [14], regeneration [15], tight junction (TJ) formation [16] and epithelial–mesenchymal transition (EMT) [17]. The presence of mutated CFTR is associated with the dysregulation of differentiation and repair, eventually leading to cancer [18], that was described as “a wound that does not heal” [19]. Other alterations associated with CFTR loss/dysfunction are implicated in the maintenance of apical-basal polarity of the airway epithelium and cytoskeletal organization. The barrier function of the airway epithelium is guaranteed by integrity by the apical junction complexes (AJC), consisting of occluding TJs, anchoring adherens junctions (AJs), desmosomes, and gap junctions (GJs). It is well described that mislocalization of TJ proteins, disorganized actin cytoskeleton and lack of actin stress fibers are present in CF cells [20][21][22][23]. The link to these alterations was found to be the interaction of CFTR with a molecular complex, tethering CFTR to the apical side of airway epithelial cells. The scaffolding protein Na+/H+ exchanger regulatory factor isoform 1 (NHERF1) is essential to maintain CFTR at the apical location through its interaction with ezrin, an A-kinase anchoring protein, that tethers PKA in the proximity of CFTR, allowing cAMP-dependent control of chloride efflux [24][25][26][27][28][29]. On the other hand, a low transepithelial resistance (TEER), a measure of epithelial tightness, indicative of TJ disorganization, has been observed in CF bronchial epithelial cells as compared with a wt-CFTR expressing cells [14][30][31]. Castellani et al. [16] confirmed these previous results on TJ disorganization in CF cells by the lack of TJ proteins at cell-cell contacts (occludin, ZO-1, claudin 1, and JAM-1), and showed also that NHERF1 or CFTR overexpression in CFBE41o- cell (F508del homozygous) monolayers induced the reorganization of TJ proteins at the level of intercellular junctions and reduced the paracellular permeability to small solutes. Interestingly, they also found that in CFBE cells, ZO-1 and occludin were localized to the nuclei, whereas the plasma membrane signal was negligible [16], indicating that CFBE cells present a dislocation of TJ proteins at the nuclear level in basal condition. In keeping with these data, Ruan and colleagues [32] have shown that in a three-dimensional (3D) epithelial cell culture model, CFTR interacts with ZO-1 at TJ levels and keeps constrained the transcription factor ZO-1 nucleic acid binding protein (ZONAB) at the same location. Upon CFTR inhibition or knockdown, ZO-1 expression is reduced and the translocation of the transcription factor ZONAB from TJs to the nucleus is induced, and an increased proliferative activity occurs.
GJs are formed by connexins (Cxs), which assemble in the plasma membrane to form hemichannels or connexons, that dock to similar connexons on the neighboring cell allowing GJ intercellular communication (GJIC). By allowing the passage of ions and small solutes, GJs are involved in the regulation of proliferation/differentiation and the maintenance of tissue homeostasis [33][34] as well as in CFTR-related epithelial functions [35][36]. CFTR has been described to interact with Cxs to regulate their trafficking and regulation of GJIC by pro-inflammatory mediators [37]. To note, it was observed that Cx43 showed perinuclear localization in CuFi-5 cells (F508del homozygous), while Cx26 localization was unaffected, and both Cxs were at the correct position in non-CF NuLi cells [31], further demonstrating a defect in cell-to-cell contacts in CF airway epithelia.
All these observations link CFTR to epithelial tightness and thus to the modulation of epithelial differentiation and proliferation, as we shall see in the following Sections dedicated to epithelial wound repair and EMT.
Events Involved in Epithelial Repair and EMT
In principle, epithelial repair follows different types of injury. In the case of CF, the damage to the airway epithelium can be caused by respiratory infections, in particular Pseudomonas aeruginosa (Figure 1) [38] and inflammatory mediators [39]. Also bacterial products secreted by P. aeruginosa, triggered by the quorum sensing (QS) network and involved in bacterial virulence, pathogenicity, and biofilm formation, have been shown to reduce airway epithelial repair rates [40][41][42]. On the other hand, heightened production of inflammatory mediators and reductions in anti-inflammatory molecule secretion by inflammatory and epithelial cells in CF lungs play a role in injury and remodeling of respiratory epithelia [43][44][45].
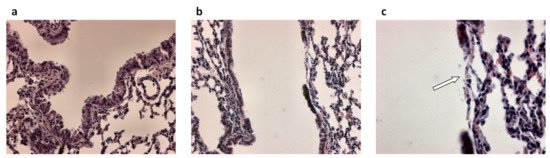
Figure 1. Airway epithelial damage in the airways following infection with P. aeruginosa. (a) Hematoxylin and eosin-stained lung section from control mice and (b,c) hematoxylin and eosin-stained lung sections from P. aeruginosa infected mice 48 h post-intratracheal instillation at a dose of 1 × 105 colony forming units. Panel c is an enlargement of panel b. Original magnification ×20 (a,b) and ×40 (c). White arrow in (c) indicates loss of the airway epithelium.
In the airways, epithelial repair follows general rules of reconstitution whose hallmarks are [46]: spreading and migration of neighboring epithelial cells using filopodial and lamellipodial extensions, a further step characterized by migration and proliferation of progenitor cells and, finally, differentiation. Recently, EMT induction, during which epithelial cells transform into mesenchymal-like cells, has been recognized as essential in physiologic and pathologic repair [47][48]. During EMT, epithelial cells lose their epithelial markers (i.e., E-cadherin) and present a migratory phenotype to re-epithelialize the wound. Fibroblast growth factor (FGF), epidermal growth factor (EGF), hepatocyte growth factor (HGF), and transforming growth factor (TGF)-β are growth factors orchestrating wound repair steps and are also involved in the initiation and regulation of the EMT [47].
The epithelium maintains its integrity through cell surface protein complexes, which contribute to form epithelial cell–cell junctions. The reorganization of the cytoskeletal architecture and polarity complexes, which result in cell shape changes, cell elongation, membrane protrusions and front–rear polarity, are essential in EMT and enable directional migration. Upon the initiation of EMT, junctions composed in the AJC are deconstructed and the junction proteins are relocalized and/or degraded [49]. A decreased claudin and occludin expression, and the diffusion of ZO-1 from cell–cell contacts has been described in early stages of EMT [50]. The findings of the nuclear localization of occludin and ZO-1 in CFBE cells [16] can be now viewed in the context of partial EMT state of CF airway epithelial cells (see below). E-cadherin—one of the main components of AJs—once cleaved and detached from the plasma membrane can be degraded [51]. Therefore, β-catenin is released from E-cadherin and act as transcription factor in the presence of WNT signaling which protects it from degradation [52]. The decrease in E-cadherin levels can even cause the accumulation of p120 catenin (also known as catenin δ1) in the nucleus where it works as transcription factor [53]. EMT initiation is also associated with disruption of desmosomes [50][51], and the decrease of Cx levels which in turn induce the loss of GJs [54]. During EMT progression, a decrease in the junction proteins expression at transcriptional level is observed, and this further stabilizes the loss of epithelial junctions [55][56]. Moreover, the apical–basal polarity, which is mediated by molecular complexes physically and functionally integrated with the cell junction architecture, is lost.
Another key role of EMT programme is to provide cells with the ability to migrate by invading through ECM. This process requires the reorganization of cytoskeleton so as to permit the dynamic elongation and directional motility [51][57][58]. Cells produce several types of membrane protrusion, which are rich of actin and facilitate cell movement and act as sensory extensions of the cytoskeleton. These projections include lamellipodia, which are sheet-like membrane protrusions, and spike-like extensions called filopodia at the edge of them [59]. Invadopodia degrade ECM by proteolytic activities, thus facilitating cell invasion [59][60]. The family of RHO GTPases is involved in these processes with RHOA promoting actin stress fibre formation and RAC1 and CDC42 promoting the formation of filopodia and lamellipodia. The conversion from apical-basal polarity (epithelial cells) to front-rear polarity (mesenchymal cells), which is one of the main aspects of EMT, involves RHO GTPases [61][62].
The repression of genes which are fundamental for the epithelial structure (e.g., E-cadherin) is counterbalanced by the activation of genes encoding proteins involved in mesenchymal adhesion such as N-cadherin (mesenchymal neural cadherin). Depending on the cell type and the extent to which cells advance through an EMT programme, cells undergoing an EMT may begin to express vimentin, to suppress cytokeratin, to shift expression of key integrins and so forth. These changes in the intermediate filament composition enable cell motility as vimentin can interact with motor proteins [63].
Remodelling of the ECM and changes to cell interactions with the ECM are essential in the initiation and progression of EMT. While acquiring a mesenchymal phenotype, epithelial cells lose their interaction with basal membrane and communicate with an inflammatory ECM.
As integrin complexes enable cells to receive ECM signals and integrate those elicited by growth factors, some epithelial integrins are down-regulated while others are activated in EMT. Some of these newly expressed integrins have key roles in EMT progression [51]. Thus, α6β4, that mediates contacts with the basement membrane, is epigenetically down-regulated [64], whereas β1 integrins increase during EMT [65][66][67][68]. During wound repair, β1-integrin subunit was found to be expressed by repairing cells on their basolateral side as well as on the apical side [69]. Kim and colleagues [65] demonstrated the crucial role of the laminin receptor α3β1 integrin in E-cadherin turnover and the remarkable crosstalk and interdependence of TGF-β signalling and β-catenin–SMAD signalling systems, in the control of EMT. Also the fibronectin receptor α5β1 integrin expression is augmented during EMT increasing cell adhesion to fibronectin, the expression of which is also activated during EMT, and promoting cell migration and protection from apoptosis. The increased expression of the β1 integrins and their ligation of collagen type I initiates a signaling cascade, leading to disassembly of the E-cadherin adhesion complex and the nuclear translocation of β-catenin, ultimately determining cell proliferation [68]. A strict connection of changes to the integrin repertoire and ligation as well as intracellular signals and increased expression of metalloproteinases (MMP2 and MMP9) has been demonstrated [49][70][71]. MMPs will enhance ECM protein degradation where integrin adhesion receptors focally interact with inflammatory ECM proteins, thus enabling invasion [51][59][72]. Other effects of MMPs related to the EMT activation are related to the trimmering of the extracellular domain of E-cadherin, thus contributing to the loss of AJs [70], and to increased SNAIL1 expression operated by increased levels of reactive oxygen species [73]. Finally, it has to be recognized that some growth factors are stored in the ECM and that localized ECM degradation may release them, such as is the case for TGF-β that is present in a latent form and is activated by MMPs and αvβ6 [74][75]. TGF-β, in turn, stimulates the expression of collagens and fibronectin, which are involved in the matrix remodeling.
EMT is induced by an interplay of soluble growth factors such as HGF, members of the TGF (e.g., TGF-β) and FGF families, insulin-like growth factor (IGFs, e.g., IGF-1), EGF as well as extracellular matrix such as collagen or hypoxic conditions. These factors activate signaling pathways leading to either expression or post-transcriptional and post-translational modification of EMT-associated transcription factors (EMT-TFs) [76][77]. Three main families of EMT-TFs have been described with the SNAI (SNAI1/Snail and SNAI2/Slug), ZEB (ZEB1 and ZEB2), and TWIST (TWIST1 and TWIST2) nuclear proteins, playing pivotal roles in the orchestration of EMT [78]. These EMT-TFs have been shown to interact with a variety of proteins involved in transcriptional regulation including proteins that function in epigenetic modification, forming together regulatory complexes.
In the airways, the final step of wound repair is the terminal differentiation which means the reconstitution of a pseudostratified epithelium with mucus-secreting cells (goblet), secretory (Club cells), ciliated cells, PNEC (pulmonary neuroendocrine cells), and basal cells. Recent studies have identified another cell type which has been called “ionocyte” and expresses higher levels of CFTR than other airway cells do [33][34]. Ionocytes derive directly from basal cells (BCs), some of which appear to function as classic multipotent stem cells, while other BCs are thought to be progenitors already committed to a ciliated or secretory fate [79][80]. As genetic (single-cell RNA sequencing) and more detailed histological and functional studies advance, not only ionocytes but also other rare cell types derived from BCs (PNEC, tuft cells, mucous ciliated cells and deuterosomal cells) are being studied during airway epithelium regeneration and repair [81][82].
The processes of epithelial repair are modulated by several growth factors, including EGF and related cytokines (TGF-α, amphiregulin, heparin-binding EGF), which through EGF receptors (EGFR), produce motility, proliferative responses, differentiation and survival [43][83][84]. MMPs and a disintegrin and metalloproteases (ADAMs) release from the cell surface EGFR ligands, including TGF-α, heparin-binding EGF (HB-EGF) and amphiregulin (AREG), that in turn can bind and activate EGFR in an autocrine or paracrine manner, while the transmembrane (pro) form may activate EGFR in adjacent cells (juxtacrine) [85]. During migration, cells attach to a provisional matrix of fibronectin and other extracellular matrix (ECM) components such as fibrin and fibrinogen. Platelet-derived growth factor (PDGF) and TGF-β act as the main modulators of ECM deposition by attracting and activating fibroblasts, in this way regulating airway repair [86][87]. Migrating epithelial cells overexpress MMPs to degrade the provisional matrix which provides novel attachment sites for lamellipodia. MMP-9 (gelatinase B), MMP-3 and MMP-11 (stromelysin 1 and 3, respectively), and MMP-7 (matrilysin), have been implicated in the matrix remodeling and acquisition of a typical epithelial-mesenchymal phenotype [88]. It has been suggested that the high expression and activation levels of MMPs can be regulated by an increase in IL-8, a pro-inflammatory cytokine [89]. TGF-β plays also a role in this remodelling process by up-regulating MMP-2 [90]. Furthermore, epithelial repair is stimulated by other growth factors, namely insulin, IGFs, HGF, keratinocyte growth factor (KGF), calcitonin gene-related peptides, and the cathelicidin LL-37 peptide. HGF and KGF acts as chemotactic and growth-stimulating factors. They also stimulate the synthesis of ECM and facilitate interactions with MMPs through specific cell receptors [91][92]. The main features of regeneration and wound repair in a non-CF airway epithelium are represented in Figure 2.
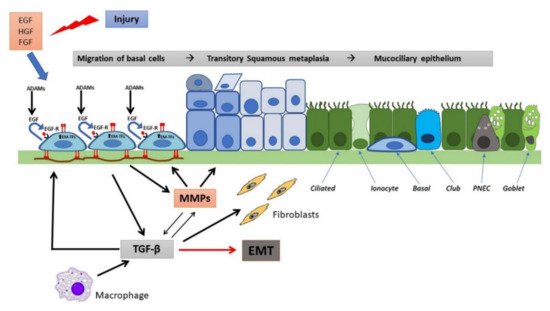
Figure 2. Wound repair and regeneration in a non-CF airway epithelium. Following injury, many cytokines and growth factors (e.g., EGF, HGF, FGF) are secreted in the wound repair microenvironment, which incite in basal cells a migratory and proliferative phenotype. Shedding of EGFR ligands (e.g., EGF) by ADAMs and binding to EGFR in an autocrine or iuxtacrine manner are key events involved in stimulation of cell migration and proliferation. Afterwards, MMP secretion by regenerating epithelial cells and subsequent TGF-β activation lead to genetic expression changes, related to EMT stimulation and activation of EMT-transcription factors (EMT-TFs). Epithelial cells and macrophages release TGF-β that induce ECM component deposition by epithelial cells and stimulate fibroblast activation, resulting in further matrix deposition. These events provoke alterations in junctional complexes and reorganization of actin cytoskeleton (not shown), modification of various integrin expression with β1-integrins increase at basal side and ectopic expression on the apical side, deposition of inflammatory ECM glycoproteins (e.g., fibronectin is shown) and its remodeling exerted by MMPs. TGF-β and MMPs enhance each other in a positive way. This process proceeds with the formation of a squamous stratified epithelium and subsequent pseudostratification and mucociliary differentiation.
2. Pathological Processes in the CF Airways
Dorothy Andersen was the first to describe the condition named “cystic fibrosis of the pancreas” and observe in 49 pediatric cases at the levels of the lungs some pathological features characteristics of CF: mild tubular dilatation of small bronchi and bronchioles, the presence of mucopurulent material in the larger bronchi and trachea with “some congestion of the underlying mucosa”, and multiple small abscesses in the smaller bronchi [93]. Her report also described the infection of airways and she observed that most of the patients in her study died of pneumonia before the age of 6 months. She and others also described squamous metaplasia of the respiratory epithelium, to be related to vitamin A deficiency and a factor in perpetuating the respiratory infections due to the absence of the protective role of mucus [94][95].
Pathological changes occurring in the airways of toddlers and older children with CF are characterized by mucopurulent plugging of small and medium size bronchioles and development of bronchiectasis, secondary to proteolysis and chondrolysis of airway support tissues [96]. The dilated airways contribute to reduced mucociliary and cough clearance and the persistence of mucus inspissation and endobronchial inflammation.
Bacterial infections and the chronic hyper-inflammatory response generated as a consequence, lead to ensuing repairing mechanisms in response to the epithelial damage [97][98][99]. Neutrophils represent the main inflammatory cell type found into the lumen of CF airways and they have been shown to contribute to the pathophysiology of CF lung disease. Indeed, these immune cells, once entered into the CF airways, produce damaging mediators (proteases, reactive oxygen species, elastase) [100] that induce epithelial apoptosis [101] and/or premature senescence [102], and herald the remodeling of the airway epithelium by upregulating mucin expression and inducing goblet cell metaplasia [103][104]. Besides the hyperactivation of the CF airway epithelium, which triggers neutrophil attraction and activation in the CF airways by producing cytokines and chemokines [105], other immune cells and mediators have been found to be involved. Elevated levels of IL-17 and IL-23 in the sputum of CF patients, and particularly in those chronically infected with P. aeruginosa, implicate a role for Th17 cells in the persistent neutrophil infiltration in CF lung disease and chronic infection with P. aeruginosa [106]. Pretreatment of immortalized CF bronchial cells (NuLi) with IL-17 determined much greater IL-8 secretion in response to an agonist of NOD1, a cytosolic innate immune receptor, and P. aeruginosa diffusible material, identifying an amplification mechanism by which CF epithelial cells may trigger the inflammatory response to bacterial ligands [107]. Furthermore, the treatment of primary CF bronchial epithelial cultures with IL-17 increased production of IL-8, IL-6 and granulocyte macrophage colony-stimulating factor, confirming a positive feedback element in CF airway inflammation involving adaptive immunity.
The structural alterations accumulating with time in CF airways include hyperplasia of goblet and basal cells [39][97][108][109], squamous metaplasia [109][110], increase in epithelial height [39][108][111], cell shedding [97][98][108][109][111], and increased thickness of reticular basement membrane (RBM) [97][112]. Airway ECM remodeling and thus structural changes follow enhanced degradation of ECM proteins such as elastin, collagen, and glycosaminoglycans. These alterations are associated with a marked and early protease/antiprotease imbalance and the release of unopposed amounts of neutrophil elastase (NE), MMPs and other proteases that are involved in tissue damage and remodeling [113][114][115][116]. In vivo data show considerable evidence of an imbalance of MMPs and their inhibitors (tissue inhibitors of MMP, TIMPs) in the CF airways with prevalence and activation of MMPs [114][117][118][119]. The action of MMPs, as well as of serine (NE) and cysteine (cathepsins) proteases, secreted by epithelial cells, macrophages and the recruited neutrophils, in the pathogenesis of CF airway disease is complex and multi-faceted, including the enhancement of mucin/mucus production and secretion, the activation of PARs (protease-activated receptors) leading to proinflammatory signaling, the trans-activation of other proteases by cleaving pro-domains and degrading cognate antiproteases, the aggravation of basic CF ion transport defects by the proteolytic degradation of CFTR and activation of ENaC, and the cleavage of various host protein substrates precipitating either activation (in the case of some proinflammatory cytokines) or inactivation (in the case of some antimicrobial peptides and surfactant proteins) [120].
Some other molecules involved directly in the remodeling of CF airways have been identified and studied. TGF-β, which has been found at elevated levels in blood (plasma) and BAL levels in CF patients and have been associated with pulmonary exacerbations [121][122], plays multiple roles in the pathogenesis of CF lung disease [123]. Besides downregulation of chloride transport in airway epithelial cells by acting negatively on CFTR and on Calcium-activated Chloride Conductance (operated by TMEM16A) [124][125][126], TGF-β signaling may drive goblet cell hyperplasia and increased mucin secretion, as indicated by studies in mouse models [127][128]. Immune and nonimmune cells, including airway epithelial cells [129][130], can secrete the latent form of TGF-β, which, upon activation orchestrates the lung immunity [131][132]. The increased RBM thickness in children with CF was found to be significantly related to BAL concentrations of TGF-β1 but unrelated to the raised levels of inflammatory cells and other cytokines [113]. Importantly, lung samples obtained from CF patients showed a significant peribronchiolar remodeling associated with prominent myofibroblast differentiation and fibrosis, i.e., TGF-β-dependent processes [129][133].
The EGFR is involved in mucin expression and secretion in the CF airways [134]. Indirect activation of MMPs and ADAMs by TNF-α stimulation can induce the EGFR pathway by evoking EGFR ligand shedding and inducing the EGFR pathway [135][136][137] in epithelial cells [138]. Both EGFR [139][140] and IL-13 receptor [141] have been associated with mucous cell metaplasia and mucin synthesis. More recently, the EGFR has been functionally coupled to ADAM17 at the level of the airway epithelium, and the EGFR/ADAM17 axis and its signaling pathway has been linked to TGF-α and AREG release and mucin expression [142]. AREG autocrine signaling affects mucus expression [143] and cytokine secretion [144], whereas its paracrine signaling has been linked to TGF-β-induced fibrosis [145]. Moreover, CF bronchial epithelial CFBE41o- cells displayed an enhanced ADAM17-mediated shedding of AREG compared with genetically identical cells with induced wt-CFTR expression and this correlated with enhanced apical presentation and phosphorylation of EGFR [146]. Further studies are necessary to clearly determine the role of EGFR/ADAM17 axis in CF, wound repair and other associated pathological hallmarks of lung disease. Nevertheless, all these changes indicate that epithelial differentiation and likely migration occurring to repair damage are somehow altered in CF airways. In the following Sections, we shall review those studies trying to elucidate the underlying mechanisms of these alterations, with particular reference to regeneration and wound repair.
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531.
- Amaral, M.D. Novel personalized therapies for cystic fibrosis: Treating the basic defect in all patients. J. Intern. Med. 2015, 277, 155–166.
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38.
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674.
- Boucher, R.C. Cystic fibrosis: A disease of vulnerability to airway surface dehydration. Trends Mol. Med. 2007, 13, 231–240.
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: Pathophysiology and therapeutic targets. Thorax 2016, 71, 284–287.
- Stutts, M.J.; Canessa, C.M.; Olsen, J.C.; Hamrick, M.; Cohn, J.A.; Rossier, B.C.; Boucher, R.C. CFTR as a cAMP-dependent regulator of sodium channel. Science 1995, 269, 847–850.
- Mall, M.; Hipper, A.; Greger, R.; Kunzelmann, K. Wild type but not deltaF508 CFTR inhibits Na+ conductance when coexpressed in Xenopus oocytes. Febs Lett. 1996, 381, 47–52.
- Donaldson, S.H.; Boucher, R.C. Sodium channels and cystic fibrosis. Chest 2007, 132, 1631–1636.
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Alaiwa, M.H.A.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113.
- Gustafsson, J.K.; Ermund, A.; Ambort, D.; Johansson, M.E.; Nilsson, H.E.; Thorell, K.; Hebert, H.; Sjovall, H.; Hansson, G.C. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J. Exp. Med. 2012, 209, 1263–1272.
- Conese, M.; Tirelli, A.S.; Alicandro, G.; Di Gioia, S.; Carbone, A.; Castellani, S.; Colombo, C. Biomarkers of Inflammation and Remodelling in Cystic Fibrosis. Clin. Immunol. Endocr. Metab. Drugs 2016, 3, 92–108.
- Larson, J.E.; Cohen, J.C. Developmental paradigm for early features of cystic fibrosis. Pediatr. Pulmonol. 2005, 40, 371–377.
- LeSimple, P.; Liao, J.; Robert, R.; Gruenert, D.C.; Hanrahan, J.W. Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J. Physiol. 2010, 588, 1195–1209.
- Hajj, R.; Lesimple, P.; Nawrocki-Raby, B.; Birembaut, P.; Puchelle, E.; Coraux, C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J. Pathol. 2007, 211, 340–350.
- Castellani, S.; Guerra, L.; Favia, M.; Di Gioia, S.; Casavola, V.; Conese, M. NHERF1 and CFTR restore tight junction organisation and function in cystic fibrosis airway epithelial cells: Role of ezrin and the RhoA/ROCK pathway. Lab. Investig. 2012, 92, 1527–1540.
- Rout-Pitt, N.; Farrow, N.; Parsons, D.; Donnelley, M. Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir. Res. 2018, 19, 136.
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What Role Does CFTR Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133.
- Byun, J.S.; Gardner, K. Wounds that will not heal: Pervasive cellular reprogramming in cancer. Am. J. Pathol. 2013, 182, 1055–1064.
- Favia, M.; Guerra, L.; Fanelli, T.; Cardone, R.A.; Monterisi, S.; Di Sole, F.; Castellani, S.; Chen, M.; Seidler, U.; Reshkin, S.J.; et al. Na+/H+ exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o-cells. Mol. Biol. Cell 2010, 21, 73–86.
- Nilsson, H.E.; Dragomir, A.; Lazorova, L.; Johannesson, M.; Roomans, G.M. CFTR and tight junctions in cultured bronchial epithelial cells. Exp. Mol. Pathol. 2010, 88, 118–127.
- Lasalvia, M.; Castellani, S.; D’Antonio, P.; Perna, G.; Carbone, A.; Colia, A.L.; Maffione, A.B.; Capozzi, V.; Conese, M. Human airway epithelial cells investigated by atomic force microscopy: A hint to cystic fibrosis epithelial pathology. Exp. Cell Res. 2016, 348, 46–55.
- Monterisi, S.; Favia, M.; Guerra, L.; Cardone, R.A.; Marzulli, D.; Reshkin, S.J.; Casavola, V.; Zaccolo, M. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J. Cell Sci. 2012, 125, 1106–1117.
- Guggino, W.B.; Stanton, B.A. New insights into cystic fibrosis: Molecular switches that regulate CFTR. Nat. Rev. Mol. Cell Biol. 2006, 7, 426–436.
- Castellani, S.; Favia, M.; Guerra, L.; Carbone, A.; Abbattiscianni, A.C.; Di Gioia, S.; Casavola, V.; Conese, M. Emerging relationship between CFTR, actin and tight junction organization in cystic fibrosis airway epithelium. Histol. Histopathol. 2017, 32, 445–459.
- Short, D.B.; Trotter, K.W.; Reczek, D.; Kreda, S.M.; Bretscher, A.; Boucher, R.C.; Stutts, M.J.; Milgram, S.L. An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J. Biol. Chem. 1998, 273, 19797–19801.
- Moyer, B.D.; Duhaime, M.; Shaw, C.; Denton, J.; Reynolds, D.; Karlson, K.H.; Pfeiffer, J.; Wang, S.; Mickle, J.E.; Milewski, M.; et al. The PDZ-interacting domain of cystic fibrosis transmembrane conductance regulator is required for functional expression in the apical plasma membrane. J. Biol. Chem. 2000, 275, 27069–27074.
- Sun, F.; Hug, M.J.; Bradbury, N.A.; Frizzell, R.A. Protein kinase A associates with cystic fibrosis transmembrane conductance regulator via an interaction with ezrin. J. Biol. Chem. 2000, 275, 14360–14366.
- Guerra, L.; Fanelli, T.; Favia, M.; Riccardi, S.M.; Busco, G.; Cardone, R.A.; Carrabino, S.; Weinman, E.J.; Reshkin, S.J.; Conese, M.; et al. Na+/H+ exchanger regulatory factor isoform 1 overexpression modulates cystic fibrosis transmembrane conductance regulator (CFTR) expression and activity in human airway 16HBE14o- cells and rescues DeltaF508 CFTR functional expression in cystic fibrosis cells. J. Biol. Chem. 2005, 280, 40925–40933.
- Weiser, N.; Molenda, N.; Urbanova, K.; Bahler, M.; Pieper, U.; Oberleithner, H.; Schillers, H. Paracellular permeability of bronchial epithelium is controlled by CFTR. Cell Physiol. Biochem. 2011, 28, 289–296.
- Molina, S.A.; Stauffer, B.; Moriarty, H.K.; Kim, A.H.; McCarty, N.A.; Koval, M. Junctional abnormalities in human airway epithelial cells expressing F508del CFTR. Am. J. Physiol. Cell Mol. Physiol. 2015, 309, L475–L487.
- Ruan, Y.C.; Wang, Y.; Da Silva, N.; Kim, B.; Diao, R.Y.; Hill, E.; Brown, D.; Chan, H.C.; Breton, S. CFTR interacts with ZO-1 to regulate tight junction assembly and epithelial differentiation through the ZONAB pathway. J. Cell Sci. 2014, 127, 4396–4408.
- Aasen, T. Connexins: Junctional and non-junctional modulators of proliferation. Cell Tissue Res. 2015, 360, 685–699.
- Vinken, M. Introduction: Connexins, pannexins and their channels as gatekeepers of organ physiology. Cell Mol. Life Sci. 2015, 72, 2775–2778.
- Huang, S.; Dudez, T.; Scerri, I.; Thomas, M.A.; Giepmans, B.N.; Suter, S.; Chanson, M. Defective activation of c-Src in cystic fibrosis airway epithelial cells results in loss of tumor necrosis factor-alpha-induced gap junction regulation. J. Biol. Chem. 2003, 278, 8326–8332.
- Losa, D.; Kohler, T.; Bellec, J.; Dudez, T.; Crespin, S.; Bacchetta, M.; Boulanger, P.; Hong, S.S.; Morel, S.; Nguyen, T.H.; et al. Pseudomonas aeruginosa-induced apoptosis in airway epithelial cells is mediated by gap junctional communication in a JNK-dependent manner. J. Immunol. 2014, 192, 4804–4812.
- Chanson, M.; Kotsias, B.A.; Peracchia, C.; O’Grady, S.M. Interactions of connexins with other membrane channels and transporters. Prog. Biophys. Mol. Biol. 2007, 94, 233–244.
- Rejman, J.; Colombo, C.; Conese, M. Engraftment of bone marrow-derived stem cells to the lung in a model of acute respiratory infection by Pseudomonas aeruginosa. Mol. Ther. 2009, 17, 1257–1265.
- Adam, D.; Roux-Delrieu, J.; Luczka, E.; Bonnomet, A.; Lesage, J.; Merol, J.C.; Polette, M.; Abely, M.; Coraux, C. Cystic fibrosis airway epithelium remodelling: Involvement of inflammation. J. Pathol. 2015, 235, 408–419.
- de Bentzmann, S.; Polette, M.; Zahm, J.-M.; Hinnrasky, J.K.C.; Bajolet, O.; Klossek, J.-M.; Filloux, A.; Lazdunski, A.; Puchelle, E. Pseudomonas aeruginosa virulence factors delay airway epithelial wound repair by altering the actin cytoskeleton and inducing overactivation of epithelial matrix metalloproteinase-2. Lab. Investig. 2000, 80, 209–219.
- Ruffin, M.; Bilodeau, C.; Maille, E.; LaFayette, S.L.; McKay, G.A.; Trinh, N.T.; Beaudoin, T.; Desrosiers, M.Y.; Rousseau, S.; Nguyen, D.; et al. Quorum-sensing inhibition abrogates the deleterious impact of Pseudomonas aeruginosa on airway epithelial repair. FASEB J. 2016, 30, 3011–3025.
- Saint-Criq, V.; Villeret, B.; Bastaert, F.; Kheir, S.; Hatton, A.; Cazes, A.; Xing, Z.; Sermet-Gaudelus, I.; Garcia-Verdugo, I.; Edelman, A.; et al. Pseudomonas aeruginosa LasB protease impairs innate immunity in mice and humans by targeting a lung epithelial cystic fibrosis transmembrane regulator-IL-6-antimicrobial-repair pathway. Thorax 2018, 73, 49–61.
- Shute, J.; Marshall, L.; Bodey, K.; Bush, A. Growth factors in cystic fibrosis-when more is not enough. Paediatr. Respir. Rev. 2003, 4, 120–127.
- Courtney, J.M.; Ennis, M.; Elborn, J.S. Cytokines and inflammatory mediators in cystic fibrosis. J. Cyst. Fibros. 2004, 3, 223–231.
- Virella-Lowell, I.; Herlihy, J.D.; Liu, B.; Lopez, C.; Cruz, P.; Muller, C.; Baker, H.V.; Flotte, T.R. Effects of CFTR, interleukin-10, and Pseudomonas aeruginosa on gene expression profiles in a CF bronchial epithelial cell Line. Mol. Ther. 2004, 10, 562–573.
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Cell Mol. Physiol. 2010, 298, L715–L731.
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506.
- Pain, M.; Bermudez, O.; Lacoste, P.; Royer, P.J.; Botturi, K.; Tissot, A.; Brouard, S.; Eickelberg, O.; Magnan, A. Tissue remodelling in chronic bronchial diseases: From the epithelial to mesenchymal phenotype. Eur. Respir. Rev. 2014, 23, 118–130.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Huang, R.Y.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422.
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33.
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779.
- Kourtidis, A.; Ngok, S.P.; Anastasiadis, P.Z. p120 catenin: An essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Prog. Mol. Biol. Transl. Sci. 2013, 116, 409–432.
- Bax, N.A.; Pijnappels, D.A.; van Oorschot, A.A.; Winter, E.M.; de Vries, A.A.; van Tuyn, J.; Braun, J.; Maas, S.; Schalij, M.J.; Atsma, D.E.; et al. Epithelial-to-mesenchymal transformation alters electrical conductivity of human epicardial cells. J. Cell Mol. Med. 2011, 15, 2675–2683.
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428.
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110.
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142.
- Yilmaz, M.; Christofori, G. Mechanisms of motility in metastasizing cells. Mol. Cancer Res. 2010, 8, 629–642.
- McNiven, M.A. Breaking away: Matrix remodeling from the leading edge. Trends Cell Biol. 2013, 23, 16–21.
- Ridley, A.J. Life at the leading edge. Cell 2011, 145, 1012–1022.
- Nelson, W.J. Remodeling epithelial cell organization: Transitions between front-rear and apical-basal polarity. Cold Spring Harb. Perspect. Biol. 2009, 1, a000513.
- Godde, N.J.; Galea, R.C.; Elsum, I.A.; Humbert, P.O. Cell polarity in motion: Redefining mammary tissue organization through EMT and cell polarity transitions. J. Mammary Gland. Biol. Neoplasia 2010, 15, 149–168.
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851.
- Yang, X.; Pursell, B.; Lu, S.; Chang, T.K.; Mercurio, A.M. Regulation of beta 4-integrin expression by epigenetic modifications in the mammary gland and during the epithelial-to-mesenchymal transition. J. Cell Sci. 2009, 122, 2473–2480.
- Kim, Y.; Kugler, M.C.; Wei, Y.; Kim, K.K.; Li, X.; Brumwell, A.N.; Chapman, H.A. Integrin alpha3beta1-dependent beta-catenin phosphorylation links epithelial Smad signaling to cell contacts. J. Cell Biol. 2009, 184, 309–322.
- Maschler, S.; Wirl, G.; Spring, H.; Bredow, D.V.; Sordat, I.; Beug, H.; Reichmann, E. Tumor cell invasiveness correlates with changes in integrin expression and localization. Oncogene 2005, 24, 2032–2041.
- Mise, N.; Savai, R.; Yu, H.; Schwarz, J.; Kaminski, N.; Eickelberg, O. Zyxin is a transforming growth factor-beta (TGF-beta)/Smad3 target gene that regulates lung cancer cell motility via integrin alpha5beta1. J. Biol. Chem. 2012, 287, 31393–31405.
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006, 66, 4662–4671.
- Herard, A.L.; Pierrot, D.; Hinnrasky, J.; Kaplan, H.; Sheppard, D.; Puchelle, E.; Zahm, J.M. Fibronectin and its alpha 5 beta 1-integrin receptor are involved in the wound-repair process of airway epithelium. Am. J. Physiol. 1996, 271, L726–L733.
- Nistico, P.; Bissell, M.J.; Radisky, D.C. Epithelial-mesenchymal transition: General principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb. Perspect. Biol. 2012, 4, a011908.
- Munshi, H.G.; Stack, M.S. Reciprocal interactions between adhesion receptor signaling and MMP regulation. Cancer Metastasis Rev. 2006, 25, 45–56.
- Shah, P.P.; Fong, M.Y.; Kakar, S.S. PTTG induces EMT through integrin alphaVbeta3-focal adhesion kinase signaling in lung cancer cells. Oncogene 2012, 31, 3124–3135.
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127.
- Sheppard, D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. 2005, 24, 395–402.
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2019, 7, 135.
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7–E8.
- Valles, A.M.; Boyer, B.; Badet, J.; Tucker, G.C.; Barritault, D.; Thiery, J.P. Acidic fibroblast growth factor is a modulator of epithelial plasticity in a rat bladder carcinoma cell line. Proc. Natl. Acad. Sci. USA 1990, 87, 1124–1128.
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036.
- Pardo-Saganta, A.; Law, B.M.; Tata, P.R.; Villoria, J.; Saez, B.; Mou, H.; Zhao, R.; Rajagopal, J. Injury induces direct lineage segregation of functionally distinct airway basal stem/progenitor cell subpopulations. Cell Stem Cell 2015, 16, 184–197.
- Watson, J.K.; Rulands, S.; Wilkinson, A.C.; Wuidart, A.; Ousset, M.; Van Keymeulen, A.; Gottgens, B.; Blanpain, C.; Simons, B.D.; Rawlins, E.L. Clonal Dynamics Reveal Two Distinct Populations of Basal Cells in Slow-Turnover Airway Epithelium. Cell Rep. 2015, 12, 90–101.
- Barbry, P.; Cavard, A.; Chanson, M.; Jaffe, A.B.; Plasschaert, L.W. Regeneration of airway epithelial cells to study rare cell states in cystic fibrosis. J. Cyst. Fibros. 2020, 19 (Suppl 1), S42–S46.
- Zaragosi, L.E.; Deprez, M.; Barbry, P. Using single-cell RNA sequencing to unravel cell lineage relationships in the respiratory tract. Biochem. Soc. Trans. 2020, 48, 327–336.
- Ingram, J.L.; Bonner, J.C. EGF and PDGF receptor tyrosine kinases as therapeutic targets for chronic lung diseases. Curr. Mol. Med. 2006, 6, 409–421.
- Knight, D. Epithelium-fibroblast interactions in response to airway inflammation. Immunol. Cell Biol. 2001, 79, 160–164.
- Gee, J.M.; Knowlden, J.M. ADAM metalloproteases and EGFR signalling. Breast Cancer Res. 2003, 5, 223–224.
- Branchett, W.J.; Lloyd, C.M. Regulatory cytokine function in the respiratory tract. Mucosal Immunol. 2019, 12, 589–600.
- Bonner, J.C. Mesenchymal cell survival in airway and interstitial pulmonary fibrosis. Fibrogenesis Tissue Repair 2010, 3, 15.
- Puchelle, E.; Zahm, J.M.; Tournier, J.M.; Coraux, C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 726–733.
- Coraux, C.; Martinella-Catusse, C.; Nawrocki-Raby, B.; Hajj, R.; Burlet, H.; Escotte, S.; Laplace, V.; Birembaut, P.; Puchelle, E. Differential expression of matrix metalloproteinases and interleukin-8 during regeneration of human airway epithelium in vivo. J. Pathol. 2005, 206, 160–169.
- Lechapt-Zalcman, E.; Pruliere-Escabasse, V.; Advenier, D.; Galiacy, S.; Charriere-Bertrand, C.; Coste, A.; Harf, A.; d’Ortho, M.P.; Escudier, E. Transforming growth factor-beta1 increases airway wound repair via MMP-2 upregulation: A new pathway for epithelial wound repair? Am. J. Physiol. Cell Mol. Physiol. 2006, 290, L1277–L1282.
- Spurzem, J.R.; Gupta, J.; Veys, T.; Kneifl, K.R.; Rennard, S.I.; Wyatt, T.A. Activation of protein kinase A accelerates bovine bronchial epithelial cell migration. Am. J. Physiol. Cell Mol. Physiol. 2002, 282, L1108–L1116.
- Waters, C.M.; Savla, U. Keratinocyte growth factor accelerates wound closure in airway epithelium during cyclic mechanical strain. J. Cell Physiol. 1999, 181, 424–432.
- Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathological study. Am. J. Dis. Child. 1938, 56, 344–399.
- Andersen, D.H. Cystic fibrosis of the pancreas, vitamin A deficiency and bronchiectasis. J. Pediatr. 1939, 15, 763–771.
- Blackfan, K.D.; Wolbach, S.B. Vitamin A deficiency in infants. J. Pediatr. 1933, 3, 679–706.
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951.
- Hubeau, C.; Lorenzato, M.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Gaillard, D. Quantitative analysis of inflammatory cells infiltrating the cystic fibrosis airway mucosa. Clin. Exp. Immunol. 2001, 124, 69–76.
- Dovey, M.; Wisseman, C.L.; Roggli, V.L.; Roomans, G.M.; Shelburne, J.D.; Spock, A. Ultrastructural morphology of the lung in cystic fibrosis. J. Submicrosc. Cytol. Pathol. 1989, 21, 521–534.
- Carson, J.L.; Collier, A.M.; Gambling, T.M.; Knowles, M.R.; Boucher, R.C. Ultrastructure of airway epithelial cell membranes among patients with cystic fibrosis. Hum. Pathol. 1990, 21, 640–647.
- Conese, M.; Castellani, S.; D’Oria, S.; di Gioia, S.; Montemurro, P. Role of Neutrophils in Cystic Fibrosis Lung Disease. In Role of Neutrophils in Disease Pathogenesis; Khajah, M.A., Ed.; IntechOpen Limited: London, UK, 2017; pp. 119–141.
- Suzuki, T.; Yamashita, C.; Zemans, R.L.; Briones, N.; Van Linden, A.; Downey, G.P. Leukocyte elastase induces lung epithelial apoptosis via a PAR-1-, NF-kappaB-, and p53-dependent pathway. Am. J. Respir. Cell Mol. Biol. 2009, 41, 742–755.
- Fischer, B.M.; Wong, J.K.; Degan, S.; Kummarapurugu, A.B.; Zheng, S.; Haridass, P.; Voynow, J.A. Increased expression of senescence markers in cystic fibrosis airways. Am. J. Physiol. Cell Mol. Physiol. 2013, 304, L394–L400.
- Voynow, J.A.; Fischer, B.M.; Malarkey, D.E.; Burch, L.H.; Wong, T.; Longphre, M.; Ho, S.B.; Foster, W.M. Neutrophil elastase induces mucus cell metaplasia in mouse lung. Am. J. Physiol. Cell Mol. Physiol. 2004, 287, L1293–L1302.
- Park, J.A.; Sharif, A.S.; Shiomi, T.; Kobzik, L.; Kasahara, D.I.; Tschumperlin, D.J.; Voynow, J.; Drazen, J.M. Human neutrophil elastase-mediated goblet cell metaplasia is attenuated in TACE-deficient mice. Am. J. Physiol. Cell Mol. Physiol. 2013, 304, L701–L707.
- De Rose, V.; Molloy, K.; Gohy, S.; Pilette, C.; Greene, C.M. Airway Epithelium Dysfunction in Cystic Fibrosis and COPD. Mediat. Inflamm. 2018, 2018, 1309746.
- Decraene, A.; Willems-Widyastuti, A.; Kasran, A.; De Boeck, K.; Bullens, D.M.; Dupont, L.J. Elevated expression of both mRNA and protein levels of IL-17A in sputum of stable Cystic Fibrosis patients. Respir. Res. 2010, 11, 177.
- Roussel, L.; Rousseau, S. IL-17 primes airway epithelial cells lacking functional Cystic Fibrosis Transmembrane conductance Regulator (CFTR) to increase NOD1 responses. Biochem. Biophys. Res. Commun. 2010, 391, 505–509.
- Voynow, J.A.; Fischer, B.M.; Roberts, B.C.; Proia, A.D. Basal-like cells constitute the proliferating cell population in cystic fibrosis airways. Am. J. Respir. Crit. Care Med. 2005, 172, 1013–1018.
- Leigh, M.W.; Kylander, J.E.; Yankaskas, J.R.; Boucher, R.C. Cell proliferation in bronchial epithelium and submucosal glands of cystic fibrosis patients. Am. J. Respir. Cell Mol. Biol. 1995, 12, 605–612.
- Piorunek, T.; Marszalek, A.; Biczysko, W.; Gozdzik, J.; Cofta, S.; Seget, M. Correlation between the stage of cystic fibrosis and the level of morphological changes in adult patients. J. Physiol. Pharm. 2008, 59 (Suppl 6), 565–572.
- Tiddens, H.A.; Koopman, L.P.; Lambert, R.K.; Elliott, W.M.; Hop, W.C.; van der Mark, T.W.; de Boer, W.J.; de Jongste, J.C. Cartilaginous airway wall dimensions and airway resistance in cystic fibrosis lungs. Eur. Respir. J. 2000, 15, 735–742.
- Durieu, I.; Peyrol, S.; Gindre, D.; Bellon, G.; Durand, D.V.; Pacheco, Y. Subepithelial fibrosis and degradation of the bronchial extracellular matrix in cystic fibrosis. Am. J. Respir. Crit. Care Med. 1998, 158, 580–588.
- Hilliard, T.N.; Regamey, N.; Shute, J.K.; Nicholson, A.G.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling in children with cystic fibrosis. Thorax 2007, 62, 1074–1080.
- Bruce, M.C.; Poncz, L.; Klinger, J.D.; Stern, R.C.; Tomashefski, J.F., Jr.; Dearborn, D.G. Biochemical and pathologic evidence for proteolytic destruction of lung connective tissue in cystic fibrosis. Am. Rev. Respir. Dis. 1985, 132, 529–535.
- Regamey, N.; Jeffery, P.K.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax 2011, 66, 624–629.
- Iosifidis, T.; Garratt, L.W.; Coombe, D.R.; Knight, D.A.; Stick, S.M.; Kicic, A. Airway epithelial repair in health and disease: Orchestrator or simply a player? Respirology 2016, 21, 438–448.
- Gaggar, A.; Hector, A.; Bratcher, P.E.; Mall, M.A.; Griese, M.; Hartl, D. The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur. Respir. J. 2011, 38, 721–727.
- Sagel, S.D.; Kapsner, R.K.; Osberg, I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr. Pulmonol. 2005, 39, 224–232.
- Garratt, L.W.; Sutanto, E.N.; Ling, K.M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Kicic-Starcevich, E.; Knight, D.A.; Ranganathan, S.; et al. Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur. Respir. J. 2015, 46, 384–394.
- McKelvey, M.C.; Weldon, S.; McAuley, D.F.; Mall, M.A.; Taggart, C.C. Targeting Proteases in Cystic Fibrosis Lung Disease. Paradigms, Progress, and Potential. Am. J. Respir. Crit. Care Med. 2020, 201, 141–147.
- Harris, W.T.; Muhlebach, M.S.; Oster, R.A.; Knowles, M.R.; Clancy, J.P.; Noah, T.L. Plasma TGF-beta(1) in pediatric cystic fibrosis: Potential biomarker of lung disease and response to therapy. Pediatr. Pulmonol. 2011, 46, 688–695.
- Harris, W.T.; Muhlebach, M.S.; Oster, R.A.; Knowles, M.R.; Noah, T.L. Transforming growth factor-beta(1) in bronchoalveolar lavage fluid from children with cystic fibrosis. Pediatr. Pulmonol. 2009, 44, 1057–1064.
- Kramer, E.L.; Clancy, J.P. TGFbeta as a therapeutic target in cystic fibrosis. Expert Opin. Targets 2018, 22, 177–189.
- Sun, H.; Harris, W.T.; Kortyka, S.; Kotha, K.; Ostmann, A.J.; Rezayat, A.; Sridharan, A.; Sanders, Y.; Naren, A.P.; Clancy, J.P. Tgf-beta downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS ONE 2014, 9, e106842.
- Snodgrass, S.M.; Cihil, K.M.; Cornuet, P.K.; Myerburg, M.M.; Swiatecka-Urban, A. Tgf-beta1 inhibits Cftr biogenesis and prevents functional rescue of DeltaF508-Cftr in primary differentiated human bronchial epithelial cells. PLoS ONE 2013, 8, e63167.
- Howe, K.L.; Wang, A.; Hunter, M.M.; Stanton, B.A.; McKay, D.M. TGFbeta down-regulation of the CFTR: A means to limit epithelial chloride secretion. Exp. Cell Res. 2004, 298, 473–484.
- Ouyang, Y.; Miyata, M.; Hatsushika, K.; Ohnuma, Y.; Katoh, R.; Ogawa, H.; Okumura, K.; Masuyama, K.; Nakao, A. TGF-beta signaling may play a role in the development of goblet cell hyperplasia in a mouse model of allergic rhinitis. Allergol. Int. 2010, 59, 313–319.
- Le, A.V.; Cho, J.Y.; Miller, M.; McElwain, S.; Golgotiu, K.; Broide, D.H. Inhibition of allergen-induced airway remodeling in Smad 3-deficient mice. J. Immunol. 2007, 178, 7310–7316.
- Harris, W.T.; Kelly, D.R.; Zhou, Y.; Wang, D.; MacEwen, M.; Hagood, J.S.; Clancy, J.P.; Ambalavanan, N.; Sorscher, E.J. Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PLoS ONE 2013, 8, e70196.
- Nicola, T.; Kabir, F.L.; Coric, T.; Wall, S.B.; Zhang, W.; James, M.; MacEwen, M.; Ren, C.; Halloran, B.; Ambalavanan, N.; et al. CFTR dysfunction increases endoglin and TGF-beta signaling in airway epithelia. Physiol. Rep. 2019, 7, e13977.
- Denney, L.; Byrne, A.J.; Shea, T.J.; Buckley, J.S.; Pease, J.E.; Herledan, G.M.; Walker, S.A.; Gregory, L.G.; Lloyd, C.M. Pulmonary Epithelial Cell-Derived Cytokine TGF-beta1 Is a Critical Cofactor for Enhanced Innate Lymphoid Cell Function. Immunity 2015, 43, 945–958.
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-beta: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655.
- Harris, W.T.; Boyd, J.T.; McPhail, G.L.; Brody, A.S.; Szczesniak, R.D.; Korbee, L.L.; Baker, M.L.; Clancy, J.P. Constrictive Bronchiolitis in Cystic Fibrosis Adolescents with Refractory Pulmonary Decline. Ann. Am. Thorac. Soc. 2016, 13, 2174–2183.
- Kreda, S.M.; Davis, C.W.; Rose, M.C. CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb. Perspect. Med. 2012, 2, a009589.
- Argast, G.M.; Campbell, J.S.; Brooling, J.T.; Fausto, N. Epidermal growth factor receptor transactivation mediates tumor necrosis factor-induced hepatocyte replication. J. Biol. Chem. 2004, 279, 34530–34536.
- Chen, W.N.; Woodbury, R.L.; Kathmann, L.E.; Opresko, L.K.; Zangar, R.C.; Wiley, H.S.; Thrall, B.D. Induced autocrine signaling through the epidermal growth factor receptor contributes to the response of mammary epithelial cells to tumor necrosis factor alpha. J. Biol. Chem. 2004, 279, 18488–18496.
- Zhou, L.; Yan, C.; Gieling, R.G.; Kida, Y.; Garner, W.; Li, W.; Han, Y.P. Tumor necrosis factor-alpha induced expression of matrix metalloproteinase-9 through p21-activated kinase-1. BMC Immunol. 2009, 10, 15.
- Maille, E.; Trinh, N.T.; Prive, A.; Bilodeau, C.; Bissonnette, E.; Grandvaux, N.; Brochiero, E. Regulation of normal and cystic fibrosis airway epithelial repair processes by TNF-alpha after injury. Am. J. Physiol. Cell Mol. Physiol. 2011, 301, L945–L955.
- Casalino-Matsuda, S.M.; Monzon, M.E.; Forteza, R.M. Epidermal growth factor receptor activation by epidermal growth factor mediates oxidant-induced goblet cell metaplasia in human airway epithelium. Am. J. Respir. Cell Mol. Biol. 2006, 34, 581–591.
- Takeyama, K.; Jung, B.; Shim, J.J.; Burgel, P.R.; Dao-Pick, T.; Ueki, I.F.; Protin, U.; Kroschel, P.; Nadel, J.A. Activation of epidermal growth factor receptors is responsible for mucin synthesis induced by cigarette smoke. Am. J. Physiol. Cell Mol. Physiol. 2001, 280, L165–L172.
- Atherton, H.C.; Jones, G.; Danahay, H. IL-13-induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation. Am. J. Physiol. Cell Mol. Physiol. 2003, 285, L730–L739.
- Stolarczyk, M.; Scholte, B.J. The EGFR-ADAM17 Axis in Chronic Obstructive Pulmonary Disease and Cystic Fibrosis Lung Pathology. Mediat. Inflamm. 2018, 2018, 1067134.
- Val, S.; Belade, E.; George, I.; Boczkowski, J.; Baeza-Squiban, A. Fine PM induce airway MUC5AC expression through the autocrine effect of amphiregulin. Arch. Toxicol. 2012, 86, 1851–1859.
- Chokki, M.; Mitsuhashi, H.; Kamimura, T. Metalloprotease-dependent amphiregulin release mediates tumor necrosis factor-alpha-induced IL-8 secretion in the human airway epithelial cell line NCI-H292. Life Sci. 2006, 78, 3051–3057.
- Zhou, Y.; Lee, J.Y.; Lee, C.M.; Cho, W.K.; Kang, M.J.; Koff, J.L.; Yoon, P.O.; Chae, J.; Park, H.O.; Elias, J.A.; et al. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-beta-induced pulmonary fibrosis. J. Biol. Chem. 2012, 287, 41991–42000.
- Stolarczyk, M.; Veit, G.; Schnur, A.; Veltman, M.; Lukacs, G.L.; Scholte, B.J. Extracellular oxidation in cystic fibrosis airway epithelium causes enhanced EGFR/ADAM17 activity. Am. J. Physiol. Cell Mol. Physiol. 2018, 314, L555–L568.
More
Information
Subjects:
Medicine, Research & Experimental
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
942
Revisions:
2 times
(View History)
Update Date:
05 Jul 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No