Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | MANKGOPO KGATLE | + 4764 word(s) | 4764 | 2021-04-20 09:48:34 | | | |
| 2 | Karina Chen | Meta information modification | 4764 | 2021-06-30 09:27:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kgatle, M. Immune Checkpoints in Prostate Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/11506 (accessed on 26 June 2026).
Kgatle M. Immune Checkpoints in Prostate Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/11506. Accessed June 26, 2026.
Kgatle, Mankgopo. "Immune Checkpoints in Prostate Cancer" Encyclopedia, https://encyclopedia.pub/entry/11506 (accessed June 26, 2026).
Kgatle, M. (2021, June 30). Immune Checkpoints in Prostate Cancer. In Encyclopedia. https://encyclopedia.pub/entry/11506
Kgatle, Mankgopo. "Immune Checkpoints in Prostate Cancer." Encyclopedia. Web. 30 June, 2021.
Copy Citation
Emerging research demonstrates that co-inhibitory immune checkpoints (ICs) remain the most promising immunotherapy targets in various malignancies. Nonetheless, ICIs have offered insignificant clinical benefits in the treatment of advanced prostate cancer (PCa) especially when they are used as monotherapies. Current existing PCa treatment initially offers an improved clinical outcome and overall survival (OS), however, after a while the treatment becomes resistant leading to aggressive and uncontrolled disease associated with increased mortality and morbidity.
immune checkpoints
immune checkpoints inhibitors
immunotherapy
metastatic hormone resistance prostate cancer
metastatic hormone sensitive prostate cancer
prostate cancer
peptide receptor ligand therapy
radionuclides
prostate specific membrane antigen
1. Introduction
PCa is the second most frequently diagnosed malignancy, and the sixth leading cause of cancer-related deaths in older men worldwide. In 2018, the incidence of PCa was approximately 1.3 million with a mortality rate of 360,000 [1]. There appears to be a positive correlation in the incidence, prevalence and increasing age of PCa; although the diagnosis of PCa remains undiagnosed in some men [2]. This may be owing to the asymptomatic PCa cases, Gleason score, screening tools, limited healthcare access and social awareness aspects in men especially in the rural settings. Additionally, routine screening procedures for PCa include digital rectal examination (DRE) for assessment of the prostate gland and use of serum prostate specific antigen (sPSA) [3]. However, these techniques are not perfect, i.e., a DRE is operator dependent and sPSA is not specific for PCa. sPSA can also be elevated in several benign and non-benign conditions (e.g., infections such as urinary tract infections, prostatitis and benign prostatic hypertrophy) which form part of PCa risk factors [4][5]. Other factors such as black ethnicity, PCa family history, having many children and diet have shown to have a positive correlation with increased risk of PCa development, although more studies are needed to determine the accuracy of some of these risk factors [3][5][6].
PCa is a notoriously heterogeneous cancer with 60–90% of the patients having multiple distinct cancer foci within the prostate gland at time of diagnosis [7]. The heterogeneity of PCa is also observed through its metastatic predominance for the skeleton with high risk of biochemical recurrence and persistence following treatment, and this correlates with increased morbidity and mortality. This has been illustrated in Figure 1 with 68Gallium(68Ga)-prostate-specific membrane antigen (PSMA)-Positron Emission Tomography (PET) images of two patients with metastatic PCa to the skeleton, which vary in the pattern of bone involvement and histological features. This probes for personalised and targeted therapeutic approach. Most PCa diagnoses and treatments are purely reliant on increased levels of sPSA and androgen hormone (AH), which is highly detectable in localised PCa and certain cancer cells. This makes advanced or aggressive PCa such as metastatic hormone-refractory PCa (mHRPC) with suppressed levels of PSA or AH difficult to diagnose and treat [8]. When diagnosed early especially at the local stage with well-differentiated non-metastatic disease, the 5-year relative survival rate of PCa is >95% as observed in more than 3000 patients with a new diagnosis recorded in the Norwegian PCa Registry in 2004–2005 [9]. PCa is classified into low, intermediate and high-risk groups, and this based on TNM stage that describes the amount and spread of cancer in patient’s body, Gleason score and level of PCa marker called sPSA [8][10].
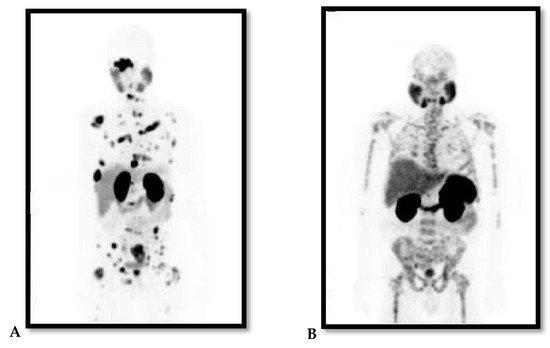
Figure 1. (A and B Maximum Projection images): 68Ga-PSMA PET images of two patients with advanced/metastatic prostate cancer to the skeleton. Note the difference in pattern of bone involvement and both are diffused skeletal demonstrating lytic and sclerotic. (A) demonstrates focal pattern but widespread lesions; (B) demonstrates lesions that are widespread in pattern interrupted with areas in the bone of no uptake within the affected bone.
2. Immune System in PCa and Advanced Stages
Chronic inflammation and immunosuppression are the hallmarks of many cancers, and are also implicated in PCa [11][12][13]. Recent studies demonstrate that IC suppression remains significantly relevant in PCa and may be efficacious in PCa patients with advanced malignancy when used in combination with other treatment modalities [12][14][15]. the ICs are immunoregulators that control the activities of T cell response by activating co-stimulatory and inhibitory immune signals. The T cells, B cells, natural killer (NK) cells, dendritic cells (DCs) and macrophages are types of immune cells, which are a network of specialised organs, tissues, cells and signalling molecules that synergise as powerful weapons to fight pathogens and cancers [12]. T cells form one of the major components of adaptive immunity that elicit responses by activating and attacking damaged cells including dendritic cells, B-cells and macrophages that have digested foreign antigens. ICs on the T cell surfaces serve as gatekeepers that control the activities of the T cell response. In normal circumstances, ICs maintain the inactive status of T cells (as naïve T cells) to prevent them from attacking and damaging the body’s own tissues or cells until they encounter specific foreign antigens [16][17][18]. Classified as either self or non-self, antigens enable the immune system to distinguish between normal interactions and antigen encounter with the foreign threat. Neoantigens are types of tumour antigen derived from mutations and tumour cells/DNA and are recognisable as non-self by the immune system. In both innate and adaptive immune responses, immune cells recognise and eliminate tumour cells in 3 principal stages [19][20]. The first stage is presentation, and in this stage the innate immune responses (neutrophils, basophils, eosinophils, and macrophages) rapidly identifies and attacks tumour cells. The resulting tumour cell death release tumour antigens, which can activate the cytotoxic T cells of the adaptive immune system. The second stage is infiltration, and it involves recruitment of immune cells by tumour antigens and other factors to the tumour site, where they invade and attacks the tumour cells. Elimination is the last stage, and here activated cytotoxic T cells recognise the tumour cells as the source of antigens and target them for elimination [19][20][21].
The immunosuppression activity of the immune cells such as Tregs and myeloid-derived suppressor cells (MDSC) is regulated by activating and inhibitory pathways. Activating pathways trigger an immune response. Inhibitory pathways such as IC pathways provide a natural counterbalance to immune activation by serving as “brakes” of the immune system that tumours usually hijack in order to shut down immune responses and protection [16][17][18][22]. This balance between inhibitory and activating pathways normally enables the immune system to attack tumour cells while sparing healthy normal cells. However, tumour cells may modulate these pathways in order to escape the detection and destruction by these immune effector cells [23][24][25]. Blockage of these pathways, especially inhibitory that tend to be implicated in various types of cancer, has shown potential therapeutic benefits by producing antitumour effects and long-term survival benefits in a broad spectrum of cancers [26][27]. This inspired the development of ICIs or blockades that led to the revolutionary treatment of many cancer types and earned both James P Allison and Tasuku Honjo a 2018 Nobel Prize in Physiology or Medicine. Patients with melanoma, bladder and kidney cancers that exhibit mismatch repair deficiency (dMMR), cyclin dependent kinase 12 (CDK12) loss and high tumour mutational burden characterised by good T cell infiltration tend to respond well to ICIs as compared to patients with PCa [28][29]. PCa is generally immunologically “cold” associated with low tumour mutational burden around tumour microenvironment and enriched with poor T cell infiltration and myeloid cells that are immunosuppressive [30][31]. However, the use of double- instead of single-agent monotherapy of ICIs or combination of single-agent monotherapy ICIs with other PCa treatment including the PCa vaccine sipuleucel-T tend to give better clinical outcomes [28][32][33]. It is, therefore, important to understand the role of inhibitory pathways in PCa to open new avenues for the development of dual combination therapeutic approaches that will favour all patients and offer a better clinical outcome. Here, we focus primarily on the recent progress in understanding inhibitory IC pathways, their inhibitors and roles in PCa treatment. These will include but is not limited to CTLA4, PD1, VISTA, IDO1, TIM3, LAG3, TIGIT, B7-H3 and B7-H4 and these are illustrated in Figure 2.
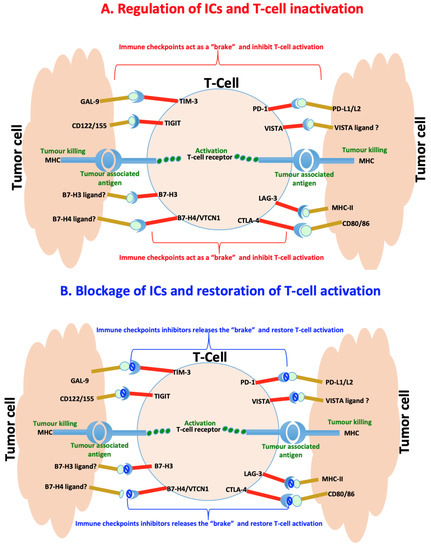
Figure 2. Immunoregulation of checkpoints in prostate cancer. (A) Immune checkpoints are usually hijacked during cancer and act as a “brake” upon binding to their ligands to slow down or inhibit cancer targeting T-cells and killing of tumor cells. (B) Counteracting the interaction of the immune checkpoints by their specific inhibitors release the “brake” and restores the T-cells functioning leading to killing of tumour cells.
3. The Immunoregulation and Inhibition of Immune Checkpoints in PCa
3.1. Cytotoxic T-lymphocyte Antigen 4
CTLA4 CTLA4 and PD1 pathways have been heavily studied, and ICIs that are under clinical studies today target these pathways or their ligands to restore antitumour responses. CTLA4 is constitutively expressed on regulatory T cells (Tregs), normal and malignant non-T cells leading to an integration network complex of positive and negative co-stimulatory signals that are required for T cell modulation. Two positive signals are required for T cell activation, and these include antigen presentation and CD28 to initiate the immune response [34]. CTLA4 and CD28 are homologous receptors both expressed by CD4+ and CD8+ T-cells which mediate opposing functions in T cell activation. They share APC expressed CD80 (B7-1) and CD86 (B7-2) as their natural ligands. Transient expression of CTLA4 occurs soon after T cell activation, resulting from ~20 times greater affinity interaction to CD80 and CD86 ligands than CD28. Thus CTLA4 outcompetes and scavenges CD80/86 away from CD28, thereby preventing CD28-mediated T cell costimulation. Subsequently, CTLA4 exerts negative inhibitory signalling to T cells by blocking CD28 co-stimulatory signal necessary for robust T cell activation and effector function. CTLA4 may also trigger trans-endocytosis and degradation of CD80 and CD86 from the cell surfaces of APC, therefore resulting in impaired costimulation via CD28-expressing T cells [35].
CTLA4 mediated T cells inhibitory signalling has been an important phenomenon implicated in various types of infections and tumours [36]. When the CTLA4 binds to its ligands, the T cells become deactivated and fail to mount the immune responses to infections and tumours. However, the blockade in CTLA4 via anti-CTLA4 ICIs is critical in disrupting the proper function of Tregs. Anti-CTLA4 binds to CTLA4 with higher affinity leading to increased accumulation, function and survival of T cells that attack tumour cells [37]. The clinical success of anti-CTLA4 ICIs was observed in advanced melanoma through the use of ipilimumab, a fully humanised antibody anti-CTLA4 monoclonal antibody (IgG) isolated from transgenic mice and produced from a hybridoma clone [38]. Ipilimumab infiltrates and represses T cell inactivation by binding to CTLA4 and preventing it from interacting with its ligands. This enables the expansion of naturally developing melanoma-specific and cytotoxic T cells that neutralise tumour cells and prevent the risk of cancer recurrence [39][40][41]. Although early phase I/II clinical trials of ipilimumab in HRPC have shown promising results with reduced cancer growth in some patients, phase III trials failed to demonstrate OS benefit [42][38][43]. Combination of ipilimumab with other therapies like docetaxel, radiotherapy, ADT, PROSTVAC, GVAX, GM-CSF resulted in PSA decline of >50% in 16% to 50% of the treated cohort, also suggesting irrelevant clinical benefit [44][45][46][47][48]. There was also no significant change in PSA doubling time when the tremelimumab was combined with ADT [49]. Poor clinical benefits of these combined therapies are likely to be attributable to the immunologically “cold” nature of tumour with relatively few tumour infiltrating T cells as mentioned earlier. However, CheckMate Trial in combination with ipilimumab and other inhibitors like nivolumab became a game changer by showing antitumour activities in both chemotherapeutic-naïve and chemotherapy-experienced HRPC patients. This was even enriched in patients with higher tumour mutational burden who benefited mostly from this treatment combination. This impressive data was compromised by observed adverse reactions (diarrhea, hypothyroidism, fatigue, skin rash, etc.) and fatalities that intercepted the use of this approach [50]. More trials with modified treatment dosage and duration approaches are ongoing to minimise these adverse reactions associated with nivolumab and ipilimumab dual-combination treatment.
3.2. Programmed Cell Death Protein 1
PD1 PD1 is an inhibitory receptor, an extended family of CTLA4/CD28 T cell regulators with two ligands including PD-L1 and PD-L2. PD1 is constitutively expressed by regulatory T cells, B cells, natural killer cells and certain myeloid cell populations, suggesting that its functional activities may be extended further than CTLA4. It is predominantly expressed by mature T cells in peripheral tissues and in the tumour microenvironment. Its pivotal roles involve balancing protective immunity and immunopathology, homeostasis and tolerance by modulating T cells response and possibly other immune cells through mechanisms that are still unknown [51]. PD1 knockout mouse exhibits significant altered immune cell development associated with autoimmune disease and congestive heart failure [52].
PD1 expression can limit protective immunity in responses to chronic infections and tumours [51]. Under normal physiological conditions, PD1 interacts with its ligands and recruits Src homology 2 (SH2) domain containing phosphatases 1/2 (SHP1/2) resulting in T cell immune suppression. Upon conventional T cells activation in response to chronic infections and various types of tumours, PD1 becomes upregulated and hijacked by some tumour cells to establish immune evasion. Immunohistochemistry studies have demonstrated that PCa cells-expressed PD-L1 are characterised by M2 macrophages, and this negatively correlates with deep changes of tumour inflammatory infiltrate composition including overexpression of PTX3, which appears to be an unfavourable prognostic marker [53]. Blockage of PD1 or PD1 ligands with pembrolizumab, nivolumab, lambrolizumab (PD1), atezolizumab and BMS-936559 (PD-L1) has demonstrated significant clinical anticancer activity by boosting T cell activation in multiple cancers including urothelial carcinoma. Pembrolizumab targets PD1 receptor by preventing it from binding to its immune-suppressing ligands, PD L1 and PD L2, therefore restoring robust T cell response that eradicate tumour cells [54]. A subset of HRPC that demonstrates dMMR pathway deficiency, a phenotype that is characterised by altered immune landscape, microsatellite instability, high mutation burden, an activated immune microenvironment, and increased PD1/PD-L1 expression on tumour and immune/stromal cells may also benefit from pembrolizumab [54][55]. McNeel et al. [56] has explored the antitumour activities of pembrolizumab and DNA vaccine encoding prostate acid phosphatase (PAP) when concurrently and sequentially combined in treating HRPC. This therapy elicited interferon-gamma (IFN-γ) secreting PAP-specific Th1-biased T cell immunity and CD8+ T cell infiltration with declined PSA only in concurrent therapy. Sadly this PSA positive response was reversed when the pembrolizumab treatment was stopped after 3 months. This suggested that the response was specifically related to the development of immune response from combination therapy of vaccination and pembrolizumab that usually targets dMMR, which were not the case in analysed patients [57].
The loss of biallaelic CDK12 is another important phenomenon and hallmark of MMR pathway deficiency in selected HRPC cases. This is usually characterised by increased gene fusions, which serve as neoantigens and promote intratumoral T cell infiltration that can potentially be targeted with pembrolizumab [28][58]. It is only 7% of HRPC patients that exhibit this genomic aberration, which means a one-size-fits-all approach that is currently being used must be reviewed and changed to specialised treatment. Developing inhibitors targeting these genomic aberrations may allow us to emulate current lung cancer model, in which the 5% to 6% of patients with non–small cell lung cancer who have an ALK rearrangement are treated with an ALK inhibitor [59][60]. This can be combined with checkpoint inhibitors and other approved HRPC treatments like sipuleucel-T to induce a favourable clinical outcome. A recent study has shown that the use of bipolar AT and enzalutamide has enhanced the response of mHRPC to anti-PD1 blockade, and this was associated with inactivation of mutations harboured by homologous recombination DNA repair genes. This suggested the therapeutic potential of IC blockade in patients with advanced PCa especially following immune activation [61]. Sena et al. [62] recently demonstrated a high clinical response rate in patients with deficient MMR after treatment with anti-PD1 pembrolizumab. Pembrolizumab resulted in prolonged progression free survival, OS and density of CD8+ tumour-infiltrating lymphocytes. These were strongly associated with tumour frameshift mutations, suggesting a new biomarker of ICIs sensitivity [62]. Nonetheless, the durability of the treatment response in some patients has been reported [62].
Unfortunately, ICI monotherapy has shown very minimal anti-cancer activity in PCa patients due to various disease factors including immunologically “cold” tumour microenvironment with poorly differentiated and few tumour infiltrating T cells. Dual combination of different ICIs or combining ICI monotherapy with currently approved HRPC hormone therapies such as enzalutamide significantly improves the efficacy of ICIs. Enzalutamide is an anti-androgen that reduces prostate tumour growth in HRPC cases by preventing the AR signalling pathway and transcriptional activities that feed tumour cells with testosterone [63]. In two separate studies including the CheckMate 650, treatment of abiraterone plus prednisone pre-treated and chemotherapy naïve HRPC patients with dual combination of enzalutamide and pembrolizumab showed a better objective response rate (ORR) with reduced PSA [46][64][65]. Graff et al. [66] recently showed that adding pembrolizumab treatment in HRPC patients progressing on enzalutamide alone induced a better clinical outcome with an objective radiographic response and PSA decrease of ≥50% as compared to HRPC patients on progressing enzalutamide monotherapy. Patients whose tumours exhibited no dMMR, CDK12 loss or PD-L1 expression also benefited from treatment in this trial [66].
Dual combination of nivolumab and ipilimumab has also demonstrated an ORR of 25% in chemotherapy-naïve HRPC cohort as compared to a HRPC cohort that has undergone chemotherapy (ORR of 10%). Here, ipilimumab turned a “cold” PCa tumour to “hot” by bringing in T cells to the tumour but simultaneously activated unneeded PD-L1, and this was blocked with nivolumab that intercepted PD1/PD-L1interaction and therefore freed the T cells to attack tumour cells. Although this combination provides a better clinical outcome, poor tolerability has still been reported and warranted for further research investigation [66].
3.3. V-Domain Immunoglobulin Suppressor of T Cell Activation
VISTA is a well-established immune regulatory receptor that can also serve as a ligand [67]. It is primarily and highly expressed in tumour infiltrating lymphocytes including in microglia, leukocytes, naïve CD4+ and Foxp3+ Tregs [68][69]. Although mechanisms underlying immunosuppressive role of VISTA are yet to be determined, VISTA has been naturally upregulated in tumour microenvironment of various malignancies such as leukaemia and pancreatic cancer. It has also recently been identified as an immunotherapy target of PCa owing it to its increased expression level in response to ipilimumab [70]. Ipilimumab-mediated expression of VISTA is directly proportional to increased expression of immuno-suppressive PD1 and PD-L1 in PCa, suggesting a compensatory inhibitory IC pathway that inspires novel effective combination-treatment strategies. A combination of ipilimumab and an anti-VISTA drug may offer a better clinical outcome in PCa [70].
3.4. Indoleamine 2,3-Dioxygenase (IDO)
Mesenchymal stem cells (MSCs) are known to play a pivotal role in tissue regeneration, wound healing and immune system, and have also been implicated in PCa development. MSCs have been demonstrated to promote transformation of androgen-dependent PCa into an androgen-independent tumour [71]. The co-culturing model of MSCs with tumour infiltrating lymphocytes has demonstrated that MSCs polarised to a Th1-like phenotype that was associated with marked pro-inflammatory changes [72]. Interferon-gamma (IFN-γ) and tumour necrosis factor-alpha (TNF-α) are two important factors that were produced by activated T cells in MSC polarisation, and this was associated with an upregulation IDO1 [73]. IDO1 catalyses the first and rate-limiting step of kynurenine pathway that converts L- tryptophan into the immunosuppressive metabolite L-kynurenine. It was also found that IDO1 is activated in some antigen-presenting cells in various tumours by tumour, MSCs and innate immune cells. IDO1 suppresses CD8+ T effector cells and natural killer cells as well as increased activity of CD4+ Tregs and MDSC. This influences immune tolerance to tumour antigens and evasion from immune-mediated destruction, which are significantly associated with poor prognosis. Previously, an increased expression of IDO1 correlated with high levels of PCa candidate biomarkers AMACR A, TNF-β1 and kynurenine in a subset of PCa patients [74]. Patients with advanced PCa exhibit an increased activation and expression of IDO1 after treatment with either DNA vaccine PAP or/and anti-PD1 inhibitor pembrolizumab. There was no PSA response as confirmed by lack of PSA decline following treatment. However, an induction of specific IFNγ-secreting T cell response was observed following in vitro stimulation of peripheral blood cells with 1-methyltryptophan that inhibit IDO. Given this data, activation and expression of IDO1 appears to be an underlying mechanism of immune evasion used by PCa. Counteracting IDO1 activities has resulted in the reactivation of anticancer immune responses in animal studies with tumour models [75], suggesting that blockage of IDO1 may also represent a promising therapeutic candidate for HRPC.
3.5. T cell Immunoglobulin Domain and Mucin Domain 3 (TIM-3)
TIM-3 is a member of Ig superfamily and is highly expressed on fully differentiated Th1 lymphocytes, CD11b+ macrophages, activated T and myeloid cells. TIM-3 regulates macrophage, activates and inhibits Th1 mediated immune responses to promote immunological tolerance. Increased level of TIM-3 expression was observed on both CD4+ and CD8+ T cells in PCa patients and this correlates with a higher Gleason score (>7) and increased pre-operative PSA [76]. On the contrary, reduced TIM-3 expression was associated with poor prognosis in metastatic PCa and served as a biomarker to differentiate metastatic HRPC from mHSPC, making the prognostic value of TIM-3 in PCa controversial [77].
Zhang et al. [78] has recently conducted preclinical studies in mice models with PCa tumours and demonstrated that triple therapy of streptavidin-GM-CSF surface-anchored tumour cell (anchored GM-CSF) vaccine, anti-TIM-3 and anti-PD1 antibodies in sequential pattern inhibited tumour growth and increased tumour regression rate in more than 60% of tested mice. This was significantly a better clinical outcome compared to concurrent therapy of anchored GM-CSF vaccine and PD1 inhibitors that, although induced robust antitumour activities, was ultimately associated with aggressive tumour progression and minimal regression in some mice. This supports numerous previous studies that demonstrated that TIM-3 is co-expressed on dysfunctional or exhausted T cells with PD1 as a compensatory and synergistic partner that, when co-blocked with PD1, reverses immune resistance in preclinical studies and restores anticancer T cell responses in patients with advanced cancer [79][80][81].
Galectin-9 (GAL9) is a TIM-3 ligand that belongs to the galectin family of lectins. Interaction of TIM-3 with GAL9 induces T cell dysfunction and predicts poor prognosis in patients with other solid tumours [82]. In PC-3 PCa cells, GAL9 was found to induce atypical ubiquitination leading to non-apoptotic cell death [83]. High-mobility group box 1 (HMGB1) and phosphatidylserine (PS) are two other ligands for TIM-3 with immunosuppressive roles in infections and various malignancies [84][85]. There may also be possible activation of TIM-3/GAL9 signaling pathway in PCa, and this still warrants research investigation to determine the effect of this pathway in PCa cell transformation and clinical outcome.
3.6. Lymphocyte-Activation Gene 3 (LAG-3)
LAG-3 is expressed on B cells, natural killer cells and plasmacytoid dendritic cells, but also constitutively and predominantly on activated cytotoxic T cells and Tregs. LAG-3 normalises both innate and adaptive immune responses by limiting cytokines secretion, T cell activation and proliferation leading to their exhaustion. Its main ligands are major histocompatibility complex class II (MHC-II) molecules, which are constitutively expressed on professional immune APCs. LAG-3 is structurally similar to CD4 receptor and both bind to antigen MHC-II as their canonical ligand (Figure 2a). However, LAG-3 binds MHC-II with 100-fold higher affinity than CD4 and negatively regulates proliferation, activation, and homeostasis of T cells in a similar fashion to CTLA4 and PD1 [84]. In the tumour microenvironment, LAG-3 becomes upregulated on Tregs, gathers around tumour sites and exerts immunosuppressive roles that amplify dysfunctional cytotoxic T cells and induction of deficient antitumour immune response. Enrichment of LAG-3 and CD8+ T-cells expressing tumour-infiltrating lymphocytes with better clinical outcome was observed in estrogen receptor negative breast cancer, indicating an independent prognostic value [86]. Indeed like TIM-3, LAG-3 can be co-expressed with other ICIs like PD1 as observed in preclinical mouse tumour models and cancer patients with intratumoral T-cell dysfunction that accentuates immune escape and increased tumour growth. This led to perturbed TNF and IFN-γ inflammatory signaling pathways which were restored with dual combination therapy counteracting both LAG-3 and PD1 activities [87][88][89][90][91]. Dual inhibition of LAG-3 and other ICIs synergistically increases T cell tumour anti-activities.
Upregulation of LAG-3 by tumour-specific CD4+ and CD8+ T cells in Tregs infiltrating PCa lesions has been observed. A contrary finding of a rather low LAG-3 expression was reported in various studies [18][92][93], suggesting that further research investigations are needed on this subject. LAG-3 also interacts with galectin-3 (GAL3) and liver sinusoidal endothelial cell lectin expressed on tumour cells and tumour-associated stromal cells, respectively. GAL3 was demonstrated to exert two opposite physiological roles based on its cellular localisation in PCa. Nuclear GAL3 may promote antitumour activities while cytoplasmic GAL3 may enhance tumour aggressiveness [94], and this may explain the contradictory level of LAG-3 expression status in the two studies. Significant reduction of nuclear GAL3 following its promoter hypermethylation has been observed in PCa, and this correlates with disease progression [95]. It has also been shown that activation of GAL3 in PCa cancer cells and xenograft mouse models with PCa causes the induction of T cell apoptosis, angiogenesis and bone metastases, which were pharmacologically reversed with RNA interference or interventions counteracting the activities of GAL3 [96][97][98][99]. Other studies have also shown that cleavage of GAL3 by metalloproteinase 2, 9 and PSA favors tumour progression in PCa suggesting its potential for therapeutic targeting [99][100]. Moreover, GAL3 activation upregulates the AR and its downstream target genes, hence, its implication in the resistance to enzalutamide and bicalutamide anti-AR drugs in xenograft mouse model [96][98].
3.7. T Cell Immunoreceptor with Ig and ITIM Domains (TIGIT)
TIGIT is also TIGIT is also known as CD223, WUCAM and VSRM3. It is a T cell and NK-cell expressed immune receptor that interacts with CD122 (Nectin-2), other nectins and CD155 (PVR) on DCs and macrophages to convert inhibitory signals on immune system [101][102][103]. TIGIT has a greater affinity to its ligands, enabling it to overcome its competitor CD226 to bind CD122 and CD155. Dysregulation of TIGIT allows tumour cells to upregulate CD122 and CD155 and avoid immune-mediated destruction. TIGIT functions like PD-L1 and, when blocked, it enhances T cell proliferation and function [104]. Co-inhibition of TIGIT with anti-TIGIT antibody tiragolumab and anti-PD-L1 antibody atezolizumab restored functional tumour-specific T cells leading to greater ORR and progression-free survival in patients with non-small lung cancer. Papanicolau-Sengos et al. [105] has demonstrated over-expression of CD122, CD155 and CD276 (B7-H3) that differentiated HRPC from HSPC, representing another potential target for aggressive PCa immunotherapy.
3.8. B7 Homolog 3 (B7-H3)
B7-H3 B7-H3 is also known as CD276, a member of the B7 family of the IgSF [106][107]. It is predominantly expressed on professional APCs including B cells, macrophages, DCs and a wide variety of tumour cells [108]. It is also expressed on a lower level in a broad variety of non-immune cells, suggesting additional non-immunological functions [106][107][108]. Both stimulatory and inhibitory properties have been identified but the ligand of B7-H3 has yet to be identified. Like TIGIT, B7 homolog 3 (B7-H3), a member of the B7 family of the IgSF, is similar to PD-L1. B7-H3 is upregulated in PCa where it is negatively correlated with biochemical cancer recurrence, progression and metastasis [109]. Several studies have revealed increased B7-H3 expression that correlates with clinicopathologic indicators of aggressive cancer, metastasis and poor clinical outcomes in PCa patients [110][111][112][113]. Moreover, expression of B7-H3 tends to be related to androgen signalling and immune reactivation [105][111][113].
Enoblituzumab is a humanised Fc-optimised B7-H3–targeting antibody that induces antibody-dependent cellular cytotoxicity (ADCC). Treatment with this monoagent conferred antitumour activities in both localised intermediate and high-risk PCa in Phase II clinical studies [114]. Phase I study of anti-B7-H3 bispecific antibody in PCa patients enhanced T cells activation and proliferation as well as production of cytokines and mediators (granzyme/perforin) that eliminates tumour cells, qualifying B7-H3 a potential target for PCa immunotherapy [115].
3.9. V-Set Domain-Containing T Cell Activation Inhibitor 1 (VTCN1)
VTCN1, also known as B7 Homolog 4 (B7-H4), is a glycosylated member of the B7 family that delivers costimulatory signals [116]. However, VTCN1 negatively regulates T cell-mediated immunity and potentiates immune evasion, epithelial cell transformation, proliferation, cytokine production and the development of cytotoxicity by suppressing T cell activation in the tumour microenvironment. Prostate, liver, kidney, lung, spleen, pancreas, placenta, testis and thymus are tissues that primarily express VTCN1 [33]. Upregulation of VTCN1 has been observed in various tumour tissues, and this is parallel with poor clinical and tumour aggressiveness pathological features [117][118]. In PCa, an elevated level of VTCN1 is associated with activation of genes that establish cancer stem cells (CSC), pathological high tumour stage and poor or shorter OS rate, making it a potential independent prognostic biomarker and therapeutic target [119][120]. CSC, a hallmark of advanced PCa, are a heterogeneous population of tumour cells with the capability for self-renewal, and are associated with increased motility that enables tumour invasion and metastases leading to PCa therapy resistance [121][122][123].
VTCN1 increased expression level is associated with a higher incidence of PD-L1 co-staining. Thus, therapeutic co-blockade of B7-H4 and PD-L1 could favourably alter the tumour microenvironment allowing for antigen-specific clearance of tumour cells [124]. Blockage of the interaction between VTCN1 and its receptors overcomes tumoral immune escape and correlates with increased T cells and NK cell infiltration that suppresses PCa tumour growth and metastasis [125][126].
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Bostwick, D.G.; Burke, H.B.; Djakiew, D.; Euling, S.; Ho, S.; Landolph, J.; Morrison, H.; Sonawane, B.; Shifflett, T.; Waters, D.J. Human Prostate Cancer Risk Factors. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2004, 101, 2371–2490.
- Perez-Cornago, A.; Key, T.J.; Allen, N.E.; Fensom, G.K.; Bradbury, K.E.; Martin, R.M.; Travis, R.C. Prospective Investigation of Risk Factors for Prostate Cancer in the UK Biobank Cohort Study. Br. J. Cancer 2017, 117, 1562–1571.
- Patel, A.R.; Klein, E.A. Risk Factors for Prostate Cancer. Nat. Clin. Pract. Urol. 2009, 6, 87–95.
- Kgatle, M.M.; Kalla, A.A.; Islam, M.M.; Sathekge, M.; Moorad, R. Prostate Cancer: Epigenetic Alterations, Risk Factors, and Therapy. Prostate Cancer 2016, 2016, 5653862.
- Ben-Shlomo, Y.; Evans, S.; Ibrahim, F.; Patel, B.; Anson, K.; Chinegwundoh, F.; Corbishley, C.; Dorling, D.; Thomas, B.; Gillatt, D. The Risk of Prostate Cancer amongst Black Men in the United Kingdom: The PROCESS Cohort Study. Eur. Urol. 2008, 53, 99–105.
- Oehus, A.; Kroeze, S.G.; Schmidt-Hegemann, N.; Vogel, M.M.; Kirste, S.; Becker, J.; Burger, I.A.; Derlin, T.; Bartenstein, P.; Eiber, M. Efficacy of PSMA Ligand PET-Based Radiotherapy for Recurrent Prostate Cancer After Radical Prostatectomy and Salvage Radiotherapy. BMC Cancer 2020, 20, 362.
- Mottet, N.; Bellmunt, J.; Bolla, M.; Briers, E.; Cumberbatch, M.G.; De Santis, M.; Fossati, N.; Gross, T.; Henry, A.M.; Joniau, S. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2017, 71, 618–629.
- Fosså, S.D.; Nilssen, Y.; Kvåle, R.; Hernes, E.; Axcrona, K.; Møller, B. Treatment and 5-Year Survival in Patients with Nonmetastatic Prostate Cancer: The Norwegian Experience. Urology 2014, 83, 146–153.
- Cooperberg, M.R.; Cowan, J.; Broering, J.M.; Carroll, P.R. High-Risk Prostate Cancer in the United States, 1990–2007. World J. Urol. 2008, 26, 211–218.
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical Roles of the Immune System during Cancer Development. Nat. Rev. Cancer 2006, 6, 24–37.
- Drake, C.G. Prostate Cancer as a Model for Tumour Immunotherapy. Nat. Rev. Immunol. 2010, 10, 580–593.
- Platz, E.A.; Kulac, I.; Barber, J.R.; Drake, C.G.; Joshu, C.E.; Nelson, W.G.; Lucia, M.S.; Klein, E.A.; Lippman, S.M.; Parnes, H.L.; et al. A Prospective Study of Chronic Inflammation in Benign Prostate Tissue and Risk of Prostate Cancer: Linked PCPT and SELECT Cohorts. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1549–1557.
- Ammirante, M.; Luo, J.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-Cell-Derived Lymphotoxin Promotes Castration-Resistant Prostate Cancer. Nature 2010, 464, 302–305.
- De Marzo, A.M.; Platz, E.A.; Sutcliffe, S.; Xu, J.; Grönberg, H.; Drake, C.G.; Nakai, Y.; Isaacs, W.B.; Nelson, W.G. Inflammation in Prostate Carcinogenesis. Nat. Rev. Cancer 2007, 7, 256–269.
- Fox, S.B.; Launchbury, R.; Bates, G.J.; Han, C.; Shaida, N.; Malone, P.R.; Harris, A.L.; Banham, A.H. The Number of Regulatory T Cells in Prostate Cancer is Associated with the Androgen Receptor and hypoxia-inducible Factor (HIF)-2α but Not HIF-1. Prostate 2007, 67, 623–629.
- Miller, A.M.; Lundberg, K.; Ozenci, V.; Banham, A.H.; Hellstrom, M.; Egevad, L.; Pisa, P. CD4+CD25high T Cells are Enriched in the Tumor and Peripheral Blood of Prostate Cancer Patients. J. Immunol. 2006, 177, 7398–7405.
- Sfanos, K.S.; Bruno, T.C.; Maris, C.H.; Xu, L.; Thoburn, C.J.; DeMarzo, A.M.; Meeker, A.K.; Isaacs, W.B.; Drake, C.G. Phenotypic Analysis of Prostate-Infiltrating Lymphocytes Reveals TH17 and Treg Skewing. Clin. Cancer Res. 2008, 14, 3254–3261.
- Gajewski, T.F.; Schreiber, H.; Fu, Y. Innate and Adaptive Immune Cells in the Tumor Microenvironment. Nat. Immunol. 2013, 14, 1014–1022.
- Moynihan, K.D.; Opel, C.F.; Szeto, G.L.; Tzeng, A.; Zhu, E.F.; Engreitz, J.M.; Williams, R.T.; Rakhra, K.; Zhang, M.H.; Rothschilds, A.M. Eradication of Large Established Tumors in Mice by Combination Immunotherapy that Engages Innate and Adaptive Immune Responses. Nat. Med. 2016, 22, 1402–1410.
- Avrameas, S.; Selmi, C. Natural Autoantibodies in the Physiology and Pathophysiology of the Immune System. J. Autoimmun. 2013, 41, 46–49.
- Sojka, D.K.; Huang, Y.; Fowell, D.J. Mechanisms of Regulatory T-cell suppression—A Diverse Arsenal for a Moving Target. Immunology 2008, 124, 13–22.
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982.
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214.
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461.
- Byun, D.J.; Wolchok, J.D.; Rosenberg, L.M.; Girotra, M. Cancer immunotherapy—Immune Checkpoint Blockade and Associated Endocrinopathies. Nat. Rev. Endocrinol. 2017, 13, 195–207.
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264.
- Rescigno, P.; de Bono, J.S. Immunotherapy for Lethal Prostate Cancer. Nat. Rev. Urol. 2019, 16, 69–70.
- Sedhom, R.; Antonarakis, E.S. Clinical Implications of Mismatch Repair Deficiency in Prostate Cancer. Future Oncol. 2019, 15, 2395–2411.
- Ihle, C.L.; Owens, P. Integrating the Immune Microenvironment of Prostate Cancer Induced Bone Disease. Mol. Carcinog. 2020, 59, 822–829.
- Bilusic, M.; Madan, R.A.; Gulley, J.L. Immunotherapy of Prostate Cancer: Facts and Hopes. Clin. Cancer Res. 2017, 23, 6764–6770.
- Slovin, S.F. Immunotherapy for Castration-Resistant Prostate Cancer: Has its Time Arrived? Expert Opin. Biol. Ther. 2020, 20, 481–487.
- Kim, T.J.; Koo, K.C. Current Status and Future Perspectives of Checkpoint Inhibitor Immunotherapy for Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 5484.
- Alegre, M.; Frauwirth, K.A.; Thompson, C.B. T-Cell Regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 2001, 1, 220–228.
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600–603.
- Vandenborre, K.; Van Gool, S.; Kasran, A.; Ceuppens, J.; Boogaerts, M.; Vandenberghe, P. Interaction of CTLA-4 (CD152) with CD80 Or CD86 Inhibits Human T-cell Activation. Immunology 1999, 98, 413–421.
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A Moving Target in Immunotherapy. Blood J. Am. Soc. Hematol. 2018, 131, 58–67.
- Small, E.J.; Tchekmedyian, N.S.; Rini, B.I.; Fong, L.; Lowy, I.; Allison, J.P. A Pilot Trial of CTLA-4 Blockade with Human Anti-CTLA-4 in Patients with Hormone-Refractory Prostate Cancer. Clin. Cancer Res. 2007, 13, 1810–1815.
- McDermott, D.; Haanen, J.; Chen, T.; Lorigan, P.; O’day, S.; MDX010-20 Investigators. Efficacy and Safety of Ipilimumab in Metastatic Melanoma Patients Surviving More than 2 Years Following Treatment in a Phase III Trial (MDX010-20). Ann. Oncol. 2013, 24, 2694–2698.
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723.
- Blansfield, J.A.; Beck, K.E.; Tran, K.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Topalian, S.L.; Haworth, L.R.; Levy, C.; et al. Cytotoxic T-Lymphocyte-Associated Antigen-4 Blockage can Induce Autoimmune Hypophysitis in Patients with Metastatic Melanoma and Renal Cancer. J. Immunother. 2005, 28, 593–598.
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; Van den Eertwegh Alfons, J.M.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H. Ipilimumab Versus Placebo After Radiotherapy in Patients with Metastatic Castration-Resistant Prostate Cancer that had Progressed After Docetaxel Chemotherapy (CA184-043): A Multicentre, Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2014, 15, 700–712.
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients with Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 40–47.
- Slovin, S.; Higano, C.; Hamid, O.; Tejwani, S.; Harzstark, A.; Alumkal, J.; Scher, H.; Chin, K.; Gagnier, P.; McHenry, M. Ipilimumab Alone or in Combination with Radiotherapy in Metastatic Castration-Resistant Prostate Cancer: Results from an Open-Label, Multicenter Phase I/II Study. Ann. Oncol. 2013, 24, 1813–1821.
- Tollefson, M.; Karnes, R.J.; Thompson, R.H.; Granberg, C.; Hillman, D.; Breau, R.; Allison, J.; Kwon, E.; Blute, M. 668 a Randomized Phase II Study of Ipilimumab with Androgen Ablation Compared with Androgen Ablation Alone in Patients with Advanced Prostate Cancer. J. Urol. 2010, 183, e261.
- Fong, L.; Kwek, S.S.; O’Brien, S.; Kavanagh, B.; McNeel, D.G.; Weinberg, V.; Lin, A.M.; Rosenberg, J.; Ryan, C.J.; Rini, B.I.; et al. Potentiating Endogenous Antitumor Immunity to Prostate Cancer through Combination Immunotherapy with CTLA4 Blockade and GM-CSF. Cancer Res. 2009, 69, 609–615.
- Jochems, C.; Tucker, J.A.; Tsang, K.; Madan, R.A.; Dahut, W.L.; Liewehr, D.J.; Steinberg, S.M.; Gulley, J.L.; Schlom, J. A Combination Trial of Vaccine Plus Ipilimumab in Metastatic Castration-Resistant Prostate Cancer Patients: Immune Correlates. Cancer Immunol. Immunother. 2014, 63, 407–418.
- Van den Eertwegh, A.J.M.; Versluis, J.; Van den Berg, H.P.; Santegoets, S.J.A.M.; Van Moorselaar, R.J.A.V.; Van der Sluis, T.M.; Gall, H.E.; Harding, T.C.; Jooss, K.; Lowy, I.; et al. Combined Immunotherapy with Granulocyte-Macrophage Colony-Stimulating Factor-Transduced Allogeneic Prostate Cancer Cells and Ipilimumab in Patients with Metastatic Castration-Resistant Prostate Cancer: A Phase 1 Dose-Escalation Trial. Lancet Oncol. 2012, 13, 509–517.
- McNeel, D.G.; Smith, H.A.; Eickhoff, J.C.; Lang, J.M.; Staab, M.J.; Wilding, G.; Liu, G. Phase I Trial of Tremelimumab in Combination with Short-Term Androgen Deprivation in Patients with PSA-Recurrent Prostate Cancer. Cancer Immunol. Immunother. 2012, 61, 1137–1147.
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Fléchon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the CheckMate 650 Trial. Cancer Cell 2020, 38, 489–499.e3.
- Sharpe, A.H.; Pauken, K.E. The Diverse Functions of the PD1 Inhibitory Pathway. Nat. Rev. Immunol. 2018, 18, 153.
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-Like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 1999, 11, 141–151.
- Scimeca, M.; Bonfiglio, R.; Urbano, N.; Cerroni, C.; Anemona, L.; Montanaro, M.; Fazi, S.; Schillaci, O.; Mauriello, A.; Bonanno, E. Programmed Death Ligand 1 Expression in Prostate Cancer Cells is Associated with Deep Changes of the Tumor Inflammatory Infiltrate Composition. Urol. Oncol. Semin. Orig. Inv. 2019, 37, 297.e19–297.e31.
- Syn, N.L.; Teng, M.W.; Mok, T.S.; Soo, R.A. De-Novo and Acquired Resistance to Immune Checkpoint Targeting. Lancet Oncol. 2017, 18, e731–e741.
- Mouw, K.W.; Konstantinopoulos, P.A. From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation. Br. J. Cancer 2018, 118, 933–935.
- McNeel, D.G.; Eickhoff, J.C.; Jeraj, R.; Staab, M.J.; Straus, J.; Rekoske, B.; Liu, G. DNA vaccine with pembrolizumab to elicit antitumor responses in patients with metastatic, castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2017, 35, 168.
- McNeel, D.G.; Eickhoff, J.C.; Wargowski, E.; Zahm, C.; Staab, M.J.; Straus, J.; Liu, G. Concurrent, but Not Sequential, PD-1 Blockade with a DNA Vaccine Elicits Anti-Tumor Responses in Patients with Metastatic, Castration-Resistant Prostate Cancer. Oncotarget 2018, 9, 25586–25596.
- Rescigno, P.; Gurel, B.; Pereira, R.; Crespo, M.; Rekowski, J.; Rediti, M.; Barrero, M.; Mateo, J.; Bianchini, D.; Messina, C.; et al. Characterizing CDK12-Mutated Prostate Cancers. Clin. Cancer Res. 2020, 27, 566–574.
- Schwab, R.; Petak, I.; Kollar, M.; Pinter, F.; Varkondi, E.; Kohanka, A.; Barti-Juhasz, H.; Schönleber, J.; Brauswetter, D.; Kopper, L. Major Partial Response to Crizotinib, a Dual MET/ALK Inhibitor, in a Squamous Cell Lung (SCC) Carcinoma Patient with De Novo c-MET Amplification in the Absence of ALK Rearrangement. Lung Cancer 2014, 83, 109–111.
- Du, X.; Shao, Y.; Qin, H.; Tai, Y.; Gao, H. ALK-rearrangement in non-small-cell Lung Cancer (NSCLC). Thorac. Cancer 2018, 9, 423–430.
- Markowski, M.C.; Shenderov, E.; Eisenberger, M.A.; Kachhap, S.; Pardoll, D.M.; Denmeade, S.R.; Antonarakis, E.S. Extreme Responses to Immune Checkpoint Blockade Following Bipolar Androgen Therapy and Enzalutamide in Patients with Metastatic Castration Resistant Prostate Cancer. Prostate 2020, 80, 407–411.
- Sena, L.A.; Fountain, J.; Isaacsson Velho, P.; Lim, S.J.; Wang, H.; Nizialek, E.; Rathi, N.; Nussenzveig, R.; Maughan, B.L.; Velez, M.G. Tumor Frameshift Mutation Proportion Predicts Response to Immunotherapy in Mismatch repair-deficient Prostate Cancer. Oncologist 2020, 26, e270–e278.
- Schalken, J.; Fitzpatrick, J.M. Enzalutamide: Targeting the Androgen Signalling Pathway in Metastatic castration-resistant Prostate Cancer. BJU Int. 2016, 117, 215–225.
- Graff, J.N.; Alumkal, J.J.; Drake, C.G.; Thomas, G.V.; Redmond, W.L.; Farhad, M.; Cetnar, J.P.; Ey, F.S.; Bergan, R.C.; Slottke, R.; et al. Early Evidence of Anti-PD-1 Activity in Enzalutamide-Resistant Prostate Cancer. Oncotarget 2016, 7, 52810–52817.
- Hansen, A.; Massard, C.; Ott, P.; Haas, N.; Lopez, J.; Ejadi, S.; Wallmark, J.; Keam, B.; Delord, J.; Aggarwal, R. Pembrolizumab for Advanced Prostate Adenocarcinoma: Findings of the KEYNOTE-028 Study. Ann. Oncol. 2018, 29, 1807–1813.
- Graff, J.N.; Beer, T.M.; Alumkal, J.J.; Slottke, R.E.; Redmond, W.L.; Thomas, G.V.; Thompson, R.F.; Wood, M.A.; Koguchi, Y.; Chen, Y.; et al. A Phase II Single-Arm Study of Pembrolizumab with Enzalutamide in Men with Metastatic Castration-Resistant Prostate Cancer Progressing on Enzalutamide Alone. J. Immunother. Cancer 2020, 8, e000642.
- Huang, X.; Zhang, X.; Li, E.; Zhang, G.; Wang, X.; Tang, T.; Bai, X.; Liang, T. VISTA: An Immune Regulatory Protein Checking Tumor and Immune Cells in Cancer Immunotherapy. J. Hematol. Oncol. 2020, 13, 1–13.
- Borggrewe, M.; Grit, C.; Den Dunnen, W.F.; Burm, S.M.; Bajramovic, J.J.; Noelle, R.J.; Eggen, B.J.; Laman, J.D. VISTA Expression by Microglia Decreases during Inflammation and is Differentially Regulated in CNS Diseases. Glia 2018, 66, 2645–2658.
- ElTanbouly, M.A.; Croteau, W.; Noelle, R.J.; Lines, J.L. VISTA: A Novel Immunotherapy Target for Normalizing Innate and Adaptive Immunity. Semin. Immunol. 2019, 42, 101308.
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E. VISTA is an Inhibitory Immune Checkpoint that is Increased After Ipilimumab Therapy in Patients with Prostate Cancer. Nat. Med. 2017, 23, 551.
- Cheng, J.; Yang, K.; Zhang, Q.; Yu, Y.; Meng, Q.; Mo, N.; Zhou, Y.; Yi, X.; Ma, C.; Lei, A. The Role of Mesenchymal Stem Cells in Promoting the Transformation of Androgen-Dependent Human Prostate Cancer Cells into Androgen-Independent Manner. Sci. Rep. 2016, 6, 16993.
- Jin, P.; Civini, S.; Zhao, Y.; De Giorgi, V.; Ren, J.; Sabatino, M.; Jin, J.; Wang, H.; Bedognetti, D.; Marincola, F. Direct T cell–tumour Interaction Triggers TH 1 Phenotype Activation through the Modification of the Mesenchymal Stromal Cells Transcriptional Programme. Br. J. Cancer 2014, 110, 2955–2964.
- Jin, P.; Zhao, Y.; Liu, H.; Chen, J.; Ren, J.; Jin, J.; Bedognetti, D.; Liu, S.; Wang, E.; Marincola, F. Interferon-γ and Tumor Necrosis Factor-α Polarize Bone Marrow Stromal Cells Uniformly to a Th1 Phenotype. Sci. Rep. 2016, 6, 26345.
- Feder-Mengus, C.; Wyler, S.; Hudolin, T.; Ruszat, R.; Bubendorf, L.; Chiarugi, A.; Pittelli, M.; Weber, W.P.; Bachmann, A.; Gasser, T.C. High Expression of Indoleamine 2, 3-Dioxygenase Gene in Prostate Cancer. Eur. J. Cancer 2008, 44, 2266–2275.
- Yu, P.; Steel, J.C.; Zhang, M.; Morris, J.C.; Waldmann, T.A. Simultaneous Blockade of Multiple Immune System Inhibitory Checkpoints Enhances Antitumor Activity Mediated by Interleukin-15 in a Murine Metastatic Colon Carcinoma Model. Clin. Cancer Res. 2010, 16, 6019–6028.
- Piao, Y.; Jin, X. Analysis of Tim-3 as a Therapeutic Target in Prostate Cancer. Tumor Biol. 2017, 39.
- Wu, J.; Lin, G.; Zhu, Y.; Zhang, H.; Shi, G.; Shen, Y.; Zhu, Y.; Dai, B.; Ye, D. Low TIM3 Expression Indicates Poor Prognosis of Metastatic Prostate Cancer and Acts as an Independent Predictor of Castration Resistant Status. Sci. Rep. 2017, 7, 1–9.
- Zhang, X.; Chen, H.; Li, G.; Zhou, X.; Shi, Y.; Zou, F.; Chen, Y.; Gao, J.; Yang, S.; Wu, S. Increased Tim-3 Expression on TILs during Treatment with the Anchored GM-CSF Vaccine and Anti-PD-1 Antibodies is Inversely Correlated with Response in Prostate Cancer. J. Cancer 2020, 11, 648.
- Fourcade, J.; Sun, Z.; Pagliano, O.; Chauvin, J.M.; Sander, C.; Janjic, B.; Tarhini, A.A.; Tawbi, H.A.; Kirkwood, J.M.; Moschos, S.; et al. PD-1 and Tim-3 Regulate the Expansion of Tumor Antigen-Specific CD8(+) T Cells Induced by Melanoma Vaccines. Cancer Res. 2014, 74, 1045–1055.
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 Expression is Associated with Tumor antigen–specific CD8 T Cell Dysfunction in Melanoma Patients. J. Exp. Med. 2010, 207, 2175–2186.
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 Finds its Place in the Cancer Immunotherapy Landscape. J. Immunother. Cancer. 2020, 8, e000911.
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G. Tim-3/galectin-9 Signaling Pathway Mediates T-cell Dysfunction and Predicts Poor Prognosis in Patients with Hepatitis B virus-associated Hepatocellular Carcinoma. Hepatology 2012, 56, 1342–1351.
- Itoh, A.; Nonaka, Y.; Ogawa, T.; Nakamura, T.; Nishi, N. Galectin-9 Induces Atypical Ubiquitination Leading to Cell Death in PC-3 Prostate Cancer Cells. Glycobiology 2019, 29, 22–35.
- Andrews, L.P.; Yano, H.; Vignali, D.A. Inhibitory Receptors and Ligands Beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or Backups. Nat. Immunol. 2019, 20, 1425–1434.
- Birge, R.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.; Huang, X.; Hutchins, J. Phosphatidylserine is a Global Immunosuppressive Signal in Efferocytosis, Infectious Disease, and Cancer. Cell Death Differ. 2016, 23, 962–978.
- Burugu, S.; Gao, D.; Leung, S.; Chia, S.; Nielsen, T. LAG-3 Tumor Infiltrating Lymphocytes in Breast Cancer: Clinical Correlates and Association with PD-1/PD-L1 Tumors. Ann. Oncol. 2017, 28, 2977–2984.
- Foy, S.P.; Sennino, B.; dela Cruz, T.; Cote, J.J.; Gordon, E.J.; Kemp, F.; Xavier, V.; Franzusoff, A.; Rountree, R.B.; Mandl, S.J. Poxvirus-Based Active Immunotherapy with PD-1 and LAG-3 Dual Immune Checkpoint Inhibition Overcomes Compensatory Immune Regulation, Yielding Complete Tumor Regression in Mice. PLoS ONE 2016, 11, e0150084.
- Huang, R.Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 Co-Inhibitory Molecules Collaborate to Limit CD8+ T Cell Signaling and Dampen Antitumor Immunity in a Murine Ovarian Cancer Model. Oncotarget 2015, 6, 27359–27377.
- Jing, W.; Gershan, J.A.; Weber, J.; Tlomak, D.; McOlash, L.; Sabatos-Peyton, C.; Johnson, B.D. Combined Immune Checkpoint Protein Blockade and Low Dose Whole Body Irradiation as Immunotherapy for Myeloma. J. Immunother. Cancer 2015, 3, 1–15.
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-Infiltrating NY-ESO-1-Specific CD8+ T Cells are Negatively Regulated by LAG-3 and PD-1 in Human Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880.
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-Cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927.
- Davidsson, S.; Andren, O.; Ohlson, A.; Carlsson, J.; Andersson, S.; Giunchi, F.; Rider, J.R.; Fiorentino, M. FOXP3 Regulatory T Cells in Normal Prostate Tissue, Postatrophic Hyperplasia, Prostatic Intraepithelial Neoplasia, and Tumor Histological Lesions in Men with and without Prostate Cancer. Prostate 2018, 78, 40–47.
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; De Marzo, A.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 Regulates CD8+ T Cell Accumulation and Effector Function in Murine Self- and Tumor-Tolerance Systems. J. Clin. Investig. 2007, 117, 3383–3392.
- Califice, S.; Castronovo, V.; Bracke, M.; van den Brûle, F. Dual Activities of Galectin-3 in Human Prostate Cancer: Tumor Suppression of Nuclear Galectin-3 Vs Tumor Promotion of Cytoplasmic Galectin-3. Oncogene 2004, 23, 7527–7536.
- Ahmed, H.; Banerjee, P.P.; Vasta, G.R. Differential Expression of Galectins in Normal, Benign and Malignant Prostate Epithelial Cells: Silencing of Galectin-3 Expression in Prostate Cancer by its Promoter Methylation. Biochem. Biophys. Res. Commun. 2007, 358, 241–246.
- Dondoo, T.; Fukumori, T.; Daizumoto, K.; Fukawa, T.; Kohzuki, M.; Kowada, M.; Kusuhara, Y.; Mori, H.; Nakatsuji, H.; Takahashi, M. Galectin-3 is Implicated in Tumor Progression and Resistance to Anti-Androgen Drug through Regulation of Androgen Receptor Signaling in Prostate Cancer. Anticancer Res. 2017, 37, 125–134.
- Guha, P.; Kaptan, E.; Bandyopadhyaya, G.; Kaczanowska, S.; Davila, E.; Thompson, K.; Martin, S.S.; Kalvakolanu, D.V.; Vasta, G.R.; Ahmed, H. Cod Glycopeptide with Picomolar Affinity to Galectin-3 Suppresses T-Cell Apoptosis and Prostate Cancer Metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 5052–5057.
- Martinez-Bosch, N.; Rodriguez-Vida, A.; Juanpere, N.; Lloreta, J.; Rovira, A.; Albanell, J.; Bellmunt, J.; Navarro, P. Galectins in Prostate and Bladder Cancer: Tumorigenic Roles and Clinical Opportunities. Nat. Rev. Urol. 2019, 16, 433–445.
- Wang, Y.; Nangia-Makker, P.; Tait, L.; Balan, V.; Hogan, V.; Pienta, K.J.; Raz, A. Regulation of Prostate Cancer Progression by Galectin-3. Am. J. Pathol. 2009, 174, 1515–1523.
- Ochieng, J.; Fridman, R.; Nangia-Makker, P.; Kleiner, D.E.; Liotta, L.A.; Stetler-Stevenson, W.G.; Raz, A. Galectin-3 is a Novel Substrate for Human Matrix Metalloproteinases-2 and-9. Biochemistry 1994, 33, 14109–14114.
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting Edge: TIGIT has T Cell-Intrinsic Inhibitory Functions. J. Immunol. 2011, 186, 1338–1342.
- Reches, A.; Ophir, Y.; Stein, N.; Kol, I.; Isaacson, B.; Amikam, Y.C.; Elnekave, A.; Tsukerman, P.; Brlic, P.K.; Lenac, T. Nectin4 is a Novel TIGIT Ligand which Combines Checkpoint Inhibition and Tumor Specificity. J. Immuno Ther. Cancer 2020, 8, e000266.
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat. Immunol. 2009, 10, 48.
- Chauvin, J.M.; Ka, M.; Pagliano, O.; Menna, C.; Ding, Q.; DeBlasio, R.; Sanders, C.; Hou, J.; Li, X.Y.; Ferrone, S.; et al. IL15 Stimulation with TIGIT Blockade Reverses CD155-Mediated NK-Cell Dysfunction in Melanoma. Clin. Cancer Res. 2020, 26, 5520–5533.
- Papanicolau-Sengos, A.; Yang, Y.; Pabla, S.; Lenzo, F.L.; Kato, S.; Kurzrock, R.; DePietro, P.; Nesline, M.; Conroy, J.; Glenn, S. Identification of Targets for Prostate Cancer Immunotherapy. Prostate 2019, 79, 498–505.
- Sun, M.; Richards, S.; Prasad, D.V.; Mai, X.M.; Rudensky, A.; Dong, C. Characterization of Mouse and Human B7-H3 Genes. J. Immunol. 2002, 168, 6294–6297.
- Ling, V.; Wu, P.W.; Spaulding, V.; Kieleczawa, J.; Luxenberg, D.; Carreno, B.M.; Collins, M. Duplication of Primate and Rodent B7-H3 Immunoglobulin V-and C-Like Domains: Divergent History of Functional Redundancy and Exon Loss. Genomics 2003, 82, 365–377.
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K. B7-H3: A Costimulatory Molecule for T Cell Activation and IFN-γ Production. Nat. Immunol. 2001, 2, 269–274.
- Bonk, S.; Tasdelen, P.; Kluth, M.; Hube-Magg, C.; Makrypidi-Fraune, G.; Möller, K.; Höflmayer, D.; Dwertmann Rico, S.; Büscheck, F.; Minner, S. High B7-H3 Expression is Linked to Increased Risk of Prostate Cancer Progression. Pathol. Int. 2020, 70, 733–742.
- Chavin, G.; Sheinin, Y.; Crispen, P.L.; Boorjian, S.A.; Roth, T.J.; Rangel, L.; Blute, M.L.; Sebo, T.J.; Tindall, D.J.; Kwon, E.D.; et al. Expression of Immunosuppresive B7-H3 Ligand by Hormone-Treated Prostate Cancer Tumors and Metastases. Clin. Cancer Res. 2009, 15, 2174–2180.
- Roth, T.J.; Sheinin, Y.; Lohse, C.M.; Kuntz, S.M.; Frigola, X.; Inman, B.A.; Krambeck, A.E.; McKenney, M.E.; Karnes, R.J.; Blute, M.L.; et al. B7-H3 Ligand Expression by Prostate Cancer: A Novel Marker of Prognosis and Potential Target for Therapy. Cancer Res. 2007, 67, 7893–7900.
- Zang, X.; Thompson, R.H.; Al-Ahmadie, H.A.; Serio, A.M.; Reuter, V.E.; Eastham, J.A.; Scardino, P.T.; Sharma, P.; Allison, J.P. B7-H3 and B7x are Highly Expressed in Human Prostate Cancer and Associated with Disease Spread and Poor Outcome. Proc. Natl. Acad. Sci. USA 2007, 104, 19458–19463.
- Benzon, B.; Zhao, S.; Haffner, M.; Takhar, M.; Erho, N.; Yousefi, K.; Hurley, P.; Bishop, J.; Tosoian, J.; Ghabili, K. Correlation of B7-H3 with Androgen Receptor, Immune Pathways and Poor Outcome in Prostate Cancer: An Expression-Based Analysis. Prostate Cancer Prostatic Dis. 2017, 20, 28–35.
- Shenderov, E.; Demarzo, A.; Boudadi, K.; Allaf, M.; Wang, H.; Chapman, C.; Pavlovich, C.; Bivalacqua, T.; O’Neal, T.S.; Harb, R. Phase II Neoadjuvant and Immunologic Study of B7-H3 Targeting with Enoblituzumab in Localized Intermediate-and High-Risk Prostate Cancer. J. Clin. Oncol. 2018, 36, TPS5099.
- Shankar, S.; Spira, A.I.; Strauss, J.; Liu, L.; La Motte-Mohs, R.; Wu, T.; Johnson, S.; Bonvini, E.; Moore, P.A.; Wigginton, J.M. A phase 1, open label, dose escalation study of MGD009, a humanized B7-H3 x CD3 DART protein, in combination with MGA012, an anti-PD-1 antibody, in patients with relapsed or refractory B7-H3-expressing tumors. J. Clin. Oncol. 2018, 36.
- Uemura, N.; Kondo, T. Current Advances in Esophageal Cancer Proteomics. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2015, 1854, 687–695.
- Xie, N.; Cai, J.; Zhang, L.; Zhang, P.; Shen, Y.; Yang, X.; Lu, J.; Gao, D.; Kang, Q.; Liu, L. Upregulation of B7-H4 Promotes Tumor Progression of Intrahepatic Cholangiocarcinoma. Cell Death Dis. 2017, 8, 1–13.
- Dong, L.; Xie, L.; Li, M.; Dai, H.; Wang, X.; Wang, P.; Zhang, Q.; Liu, W.; Hu, X.; Zhao, M. Downregulation of B7-H4 Suppresses Tumor Progression of Hepatocellular Carcinoma. Sci. Rep. 2019, 9, 1–12.
- Li, H.; Piao, L.; Liu, S.; Cui, Y.; Xuan, Y. B7-H4 is a Potential Prognostic Biomarker of Prostate Cancer. Exp. Mol. Pathol. 2020, 114, 104406.
- Qian, Y.; Yao, H.P.; Shen, L.; Cheng, L.F.; Zhang, L.H. Expression of B7-H4 in Prostate Cancer and its Clinical Significance. Zhejiang Da Xue Xue Bao Yi Xue Ban 2010, 39, 345–349.
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344.
- Santer, F.R.; Erb, H.H.; McNeill, R.V. Therapy Escape Mechanisms in the Malignant Prostate. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2015; pp. 133–144.
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-Associated microglia/macrophages Enhance the Invasion of Glioma Stem-Like Cells Via TGF-beta1 Signaling Pathway. J. Immunol. 2012, 189, 444–453.
- Krambeck, A.E.; Thompson, R.H.; Dong, H.; Lohse, C.M.; Park, E.S.; Kuntz, S.M.; Leibovich, B.C.; Blute, M.L.; Cheville, J.C.; Kwon, E.D. B7-H4 Expression in Renal Cell Carcinoma and Tumor Vasculature: Associations with Cancer Progression and Survival. Proc. Natl. Acad. Sci. USA 2006, 103, 10391–10396.
- Dangaj, D.; Lanitis, E.; Zhao, A.; Joshi, S.; Cheng, Y.; Sandaltzopoulos, R.; Ra, H.J.; Danet-Desnoyers, G.; Powell, D.J., Jr.; Scholler, N. Novel Recombinant Human b7-h4 Antibodies Overcome Tumoral Immune Escape to Potentiate T-Cell Antitumor Responses. Cancer Res. 2013, 73, 4820–4829.
- Jeon, H.; Vigdorovich, V.; Garrett-Thomson, S.C.; Janakiram, M.; Ramagopal, U.A.; Abadi, Y.M.; Lee, J.S.; Scandiuzzi, L.; Ohaegbulam, K.C.; Chinai, J.M. Structure and Cancer Immunotherapy of the B7 Family Member B7x. Cell Rep. 2014, 9, 1089–1098.
More
Information
Subjects:
Virology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
773
Revisions:
2 times
(View History)
Update Date:
30 Jun 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No