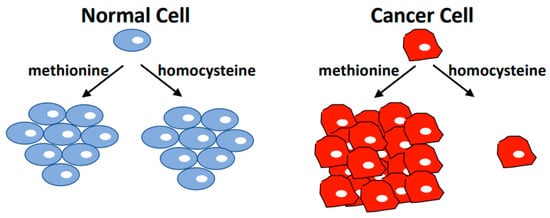
Tumorigenesis is accompanied by the reprogramming of cellular metabolism. The shift from oxidative phosphorylation to predominantly glycolytic pathways to support rapid growth is well known and is often referred to as the Warburg effect. However, other metabolic changes and acquired needs that distinguish cancer cells from normal cells have also been discovered. The dependence of cancer cells on exogenous methionine is one of them and is known as methionine dependence or the Hoffman effect. This phenomenon describes the inability of cancer cells to proliferate when methionine is replaced with its metabolic precursor, homocysteine, while proliferation of non-tumor cells is unaffected by these conditions. Surprisingly, cancer cells can readily synthesize methionine from homocysteine, so their dependency on exogenous methionine reflects a general need for altered metabolic flux through pathways linked to methionine.
1. Introduction
In 1959, Sugimura and colleagues reported a study where tumor-bearing rats were fed diets with the restriction of individual essential amino acids. Tumor growth was significantly affected by a methionine-restricted diet
[1]. The dependence of cancer cell proliferation on methionine was further highlighted in 1973 by experiments that showed leukemia cells cannot proliferate in growth media where methionine is substituted with its metabolic precursor, homocysteine
[2]. A flurry of experiments during the 70s and 80s expanded the methionine/homocysteine substitution experiments to many cell lines derived from various tumor sites. The results unequivocally established that the vast majority of cancer cells cannot proliferate when methionine is replaced with homocysteine, but non-cancer cells are indifferent to such replacement
[3][4][5][6][7]. lists cell lines with a known status of methionine dependence. This metabolic phenomenon that differentiates cancer cells from non-tumor cells is often referred to as the methionine dependence of cancer, the methionine stress sensitivity of cancer, or the Hoffman effect (). It soon became clear that the addiction of cancer cells to exogenously provided methionine is not due to their failure to synthesize methionine from homocysteine
[3][8], but is likely caused by increased demand for metabolites derived from methionine
[5][7][8][9]. While mechanisms behind the Hoffman effect are yet to be fully understood, progress has been made towards this goal.
Figure 1. The Hoffman Effect. Non-tumorigenic cell lines have the same proliferation rate in media containing methionine or media where methionine is replaced with the immediate metabolic precursor homocysteine. However, most cancer cells cannot proliferate in homocysteine medium, and induce cell cycle arrest followed by apoptosis when cultured under these conditions. Cancer cells readily synthesize methionine from homocysteine, but appear to depend on exogenously supplied methionine. This cancer-specific metabolic dependence is referred to as the methionine dependence of cancer, the methionine stress sensitivity of cancer, or simply the Hoffman effect.
Table 1. Cell lines with known growth properties in homocysteine medium.
| Cell Line |
Methionine Dependence |
Tumor Site |
| MDA-MB468 |
Yes [7][10] |
Breast |
| MCF7 |
Yes [5][6][7][10] |
Breast |
| MDA-MB361 |
Yes [7] |
Breast |
| HCC1806 |
Yes [10] |
Breast |
| HCC1143 |
Yes [10] |
Breast |
| SKBR3 |
Yes [10] |
Breast |
| BT-549 |
Yes [10] |
Breast |
| ZR-75-1 |
Yes [10] |
Breast |
| SUM-159 |
Yes [10] |
Breast |
| T47D |
Yes [10] |
Breast |
| W-256 |
Yes [3][4] |
Breast (rat) |
| MDA-MB231 |
No [1], moderate [10] |
Breast |
| HCC70 |
No [10] |
Breast |
| HCC38 |
No [10] |
Breast |
| SUM-149 |
No [10] |
Breast |
| MDA-MB231 |
No [1], moderate [10] |
Breast |
| BxPC3 |
Yes [11] |
Pancreas |
| PANC1 |
No [11] |
Pancreas |
| LoVo |
Yes [12] |
Colon |
| SK-CO-1 |
Yes [6] |
Colon |
| PC-3 |
Yes [5][6][13] |
Prostate |
| LNCaP |
Moderate [13] |
Prostate |
| DU145 |
Moderate [5][6][13] |
Prostate |
| SV80 |
Yes [3] |
Transformed fibroblast |
| HEK293T |
Yes [11] |
Transformed kidney cell |
| W18VA2 |
Yes [3] |
SV40 transformed human cells |
| J111 |
Yes [4] |
Monocytic leukemia |
| L1210 |
Yes [4] |
Lymphatic leukemia (mouse) |
| A2182 |
Yes [5][6] |
Lung |
| SK-LU-1 |
Yes [5][6] |
Lung |
| A549 |
Moderate [6] |
Lung |
| A427 |
No [5][6] |
Lung |
| J82 |
Yes [5][6] |
Bladder |
| T24 |
No [5][6] |
Bladder |
| 8387 |
Yes [5][6] |
Fibrosarcoma |
| HT1080 |
Yes [5][6] |
Fibrosarcoma |
| HOS |
Yes [5][6] |
Osteosarcoma |
| A204 |
Moderate [6] |
Rhabdomyosarcoma |
| A673 |
Yes [5][6] |
Rhabdomyosarcoma |
| SK-LMS1 |
No [5][6] |
Leiomyosarcoma |
| SK-N-SH |
Yes [5][6] |
Neuroblastoma |
| SK-N-MC |
No [6] |
Neuroblastoma |
| A375 |
Moderate [6] |
Melanoma |
| MeWo |
No [6] |
Melanoma |
| A172 |
Moderate [5][6] |
Glioblastoma |
| HeLa |
Yes [6] |
Cervical |
| A498 |
Yes [5][6] |
Kidney |
2. Methionine Metabolism
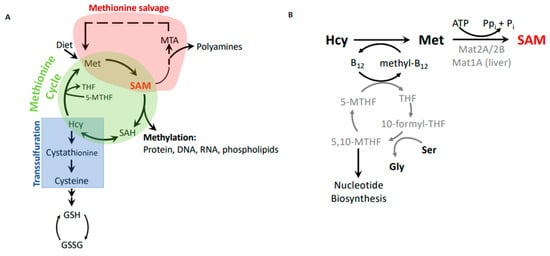
Methionine is an essential amino acid in mammals. In addition to its role as a component of proteins, methionine links to a number of important metabolic pathways that play key roles in epigenetics (S-adenosylmethionine), nuclear functions (polyamines), detoxification (glutathione), and cellular membranes (phospholipids) (). Furthermore, the methionine cycle is intimately linked with folate metabolism and thus can indirectly modulate nucleotide biosynthesis.
Figure 2. Methionine metabolism. (A) Metabolic connections between the methionine cycle, which produces methylation potential, the methionine salvage pathway, which recycles methionine from byproducts of the polyamine synthesis pathway, and the transsulfuration pathway, which generates cysteine and glutathione to combat oxidation. (B) The remethylation step of homocysteine as part of the methionine cycle and synthesis of S-adenosylmethionine (SAM).
Methionine is obtained through the diet. It is converted to the principal cellular methyl donor,
S-adenosylmethionine (SAM, also referred to as AdoMet), through the transfer of adenosine from ATP to the methionine sulfur. This reaction is catalyzed by methionine adenosyl transferases (MAT). Mat1A is the main transferase in the liver, whereas extrahepatic tissues rely on the Mat2A/Mat2B complex for SAM synthesis. Mat2A is the catalytic subunit, but binding of the regulatory subunit Mat2B modifies kinetic properties by decreasing the K
m for methionine and sensitizing the enzyme to product inhibition
[14][15][16]. SAM is used as a cofactor in most methylation reactions and provides the activated methyl group for conjugation to proteins, DNA, and lipids.
S-adenosylhomocysteine (SAH) remains after the methyl group transfer and is hydrolyzed into homocysteine and adenosine by SAH hydrolase. Homocysteine is then remethylated using 5-methyltetrahydrofolate as the methyl group donor to regenerate methionine and complete the methionine cycle (A,B). Notably, vitamin B
12 is required to transfer methyl groups from 5-methyltetrahydrofolate to remethylate homocysteine (B). In the liver and kidney, homocysteine can also be remethylated using betaine, which is derived from choline. However, most other tissues do not express the necessary enzyme, betaine–homocysteine methyltransferase
[17][18], and rely on the folate and B
12-mediated remethylation reaction.
Maintaining the necessary methylation potential in cells via generation of SAM is a major role of methionine metabolism. However, one needs to consider that SAH is a potent inhibitor of methyltransferases
[19], and the ability of cells to methylate substrates is thus not only determined by SAM abundance, but also by SAH levels. The cellular methylation potential is thus best expressed as the SAM/SAH ratio.
3. Methionine Metabolism and Cancer
Methionine metabolism has been connected to cancer on several levels. This review focuses on the Hoffman effect, which describes the dependence of cancer cells on exogenous methionine. Most cancer cells cannot proliferate in medium where methionine is replaced by homocysteine, even though they readily synthesize methionine from homocysteine (). In fact, when intracellular methionine levels were measured in breast cancer cells after they had been shifted to homocysteine medium, methionine levels remained largely constant
[8]. Non-cancer cells are indifferent to replacement of methionine with homocysteine. Differential metabolic dependencies of cancer and normal cells are often difficult to interpret, because different growth rates of cancer and normal cells can indirectly lead to distinct metabolic needs. However, the existence of several rapidly proliferating methionine-independent cancer cell lines argues against a major influence of proliferation rate ()
[5][6]. Why some cancer cells remain methionine independent is not well understood. In addition, more direct approaches have definitely excluded indirect effects from growth rate-related metabolic dependence
[7][8][20][21][22]. Most notably, when methionine-dependent cancer cells are continuously cultured in homocysteine medium, very rare cell clones can be selected that reverted to methionine independence without a change in proliferation rate
[7][8][20][21][22]. Remarkably, most of these cancer cell-derived methionine-independent clones have lost properties associated with the tumorigenic state
[8][20]. The mechanism of reversion is not known, but chromosomal alterations have been correlated with reversion to methionine independence in some systems
[21]. However, reversion of MDA-MB468 triple negative breast cancer cells seems to be an epigenetic event, because the reverted state is semi-stable and needs to be stabilized occasionally by growth in homocysteine medium
[22]. As indicated above, generation of methionine stress resistant cancer cells is usually coupled with loss of tumorigenic properties such as the ability of anchorage independent growth and proliferation in 1% serum. Conversely, oncogenic mutations in phosphoinositide 3-kinase (PI3K) and H-ras expression have been shown to promote methionine dependence
[10][23]. Whether other oncogenes induce a similar metabolic dependence has not been systematically investigated, but these experiments indicate a tight link between methionine dependence and tumorigenicity. These findings are also encouraging considering clinical implications. Treatment strategies exploiting the methionine dependence of cancer should not be easily overcome by tumors developing resistance to methionine restriction, because these cancer cell line experiments indicate that tumorigenic properties are lost when cancer cells escape methionine dependence.
Even though cancer cells readily synthesize methionine from homocysteine and show similar intracellular steady-state methionine levels as methionine-independent cells, they cannot proliferate in these conditions. There are indications of a qualitative difference between exogenous and homocysteine-derived methionine. Such a different utilization of exogenous and synthesized methionine has been noted in double-label experiments, where cells preferentially incorporated exogenous methionine
[24]. The reason for this difference is unknown. One issue to consider is that steady-state metabolite measurements do not inform us about flux through metabolic pathways and can therefore be misleading. For example, methionine levels may be kept constant at the expense of flux into downstream pathways. Indeed, tracing experiments in MDA-MB468 breast cancer cells with labelled homocysteine revealed a diversion of flux from the methionine cycle to the transsulfuration pathway (A)
[8]. These same experiments indicated the reduced synthesis of SAM, resulting in a reduced overall methylation potential reflected in a lower SAM/SAH ratio. These tracing experiments suggested that low SAM or SAM/SAH ratios are key to understanding the Hoffman effect, and that increased flux through the transsulfuration pathway may induce these changes in methylation potential. Notably, none of these metabolic effects were observed in revertant, methionine-independent MDA-MB468-R8 clones. The differential metabolic fates of homocysteine in methionine-dependent (transsulfuration) and -independent cells (mainly methionine cycle) observed in breast cancer cell systems may be a general feature of the Hoffman effect. Indeed, similar redirection of homocysteine into the transsulfuration branch is induced by oncogenic PI3K mutations, which induce a methionine-dependent phenotype when expressed in methionine-independent cells (A)
[10]. It is interesting to note that the transsulfuration pathway is largely restricted to pancreas, liver, and kidney, but is active in cancer cells
[8][25][26][27], which could contribute to the differential response of cancer cells to growth in homocysteine medium. One can speculate that higher demand for antioxidants like glutathione in cancer may require increased flux through the transsulfuration pathway. However, at least in MDA-MB468 breast cancer cells, supplementation with antioxidants cannot compensate for methionine dependence
[28].
Effects of methionine substitution with homocysteine on cellular methylation potential have been observed previously
[3], but the significance of SAM as an important mediator of the Hoffman effect became clear when SAM supplementation of homocysteine medium eliminated the growth defect of MDA-MB468 cells
[7]. Accordingly, reducing SAM synthesis directly by knockdown of MAT2A/B without affecting methionine levels resulted in a similar cell cycle arrest as observed by methionine replacement
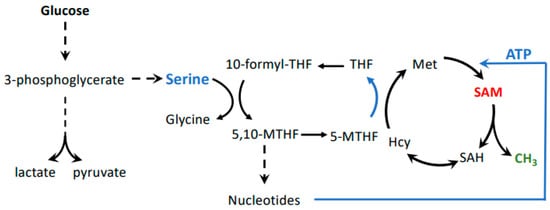
[9]. The realization that SAM is one of the key metabolites related to the methionine dependence of cancer may also connect other metabolic pathways important to cancer with the Hoffman effect. For example, the serine–glycine biosynthesis pathway is critical to maintaining cellular methylation potential (B). This pathway maintains flux through the folate cycle. Thereby, serine to glycine conversion sustains homocysteine remethylation to methionine, but also stimulates nucleotide biosynthesis, including ATP, which, together with methionine, provides the building blocks for SAM ()
[29]. Several cancer cell lines and tumors depend on exogenous serine. This is surprising because serine can usually be synthesized from glucose in sufficient amounts via the glycolysis intermediate 3-phosphoglycerate
[30][31]. However, the increased demand of cancer cells on glycolysis for energy production may divert the flux of 3-phosphoglycerate from serine synthesis to glycolysis. Serine requirement for cancer cell proliferation may thus link the Warburg and Hoffman effects by connecting glycolytic flux with SAM synthesis.
Figure 3. Possible connection between the Warburg and Hoffman effects. Glycolysis is connected to the methionine cycle through the folate cycle, whereby serine derived from 3-phosphoglycerate provides one-carbon units. This pathway is important in cancer cells because the folate cycle feeds both nucleotide and SAM biosynthesis. SAM levels are especially impacted by the folate cycle because it is important for re-methylation of homocysteine to methionine, as well as ATP synthesis. Both methionine and ATP are substrates for formation of SAM.
 +1 credit
+1 credit