 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christian Schlieker | + 2879 word(s) | 2879 | 2021-06-04 10:44:15 | | | |
| 2 | Vivi Li | Meta information modification | 2879 | 2021-06-30 04:41:23 | | |
Video Upload Options
Torsin ATPases are members of the AAA+ (ATPases associated with various cellular activities) superfamily of proteins, which participate in essential cellular processes. While AAA+ proteins are ubiquitously expressed and demonstrate distinct subcellular localizations, Torsins are the only AAA+ to reside within the nuclear envelope (NE) and endoplasmic reticulum (ER) network. Moreover, due to the absence of integral catalytic features, Torsins require the NE- and ER-specific regulatory cofactors, lamina-associated polypeptide 1 (LAP1) and luminal domain like LAP1 (LULL1), to efficiently trigger their atypical mode of ATP hydrolysis. Despite their implication in an ever-growing list of diverse processes, the specific contributions of Torsin/cofactor assemblies in maintaining normal cellular physiology remain largely enigmatic. Resolving gaps in the functional and mechanistic principles of Torsins and their cofactors are of considerable medical importance, as aberrant Torsin behavior is the principal cause of the movement disorder DYT1 early-onset dystonia.
1. Introduction
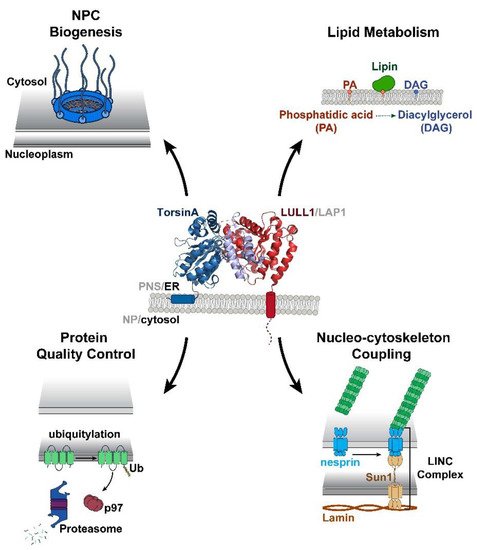
2. Structural and Biochemical Perspectives on Torsins
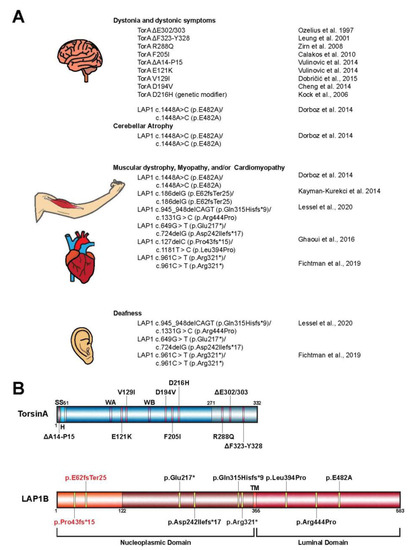
3. Torsin Assemblies and Dystonia Movement Disorders
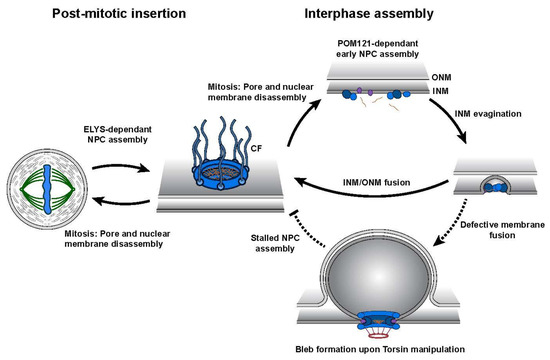
4. The Role of Torsin ATPases in Nuclear Pore Biogenesis
References
- Ozelius, L.J.; Hewett, J.W.; Page, C.E.; Bressman, S.B.; Kramer, P.L.; Shalish, C.; de Leon, D.; Brin, M.F.; Raymond, D.; Corey, D.P.; et al. The early-onset torsion dystonia gene (dyt1) encodes an atp-binding protein. Nat. Genet. 1997, 17, 40–48.
- Ozelius, L.J.; Page, C.E.; Klein, C.; Hewett, J.W.; Mineta, M.; Leung, J.; Shalish, C.; Bressman, S.B.; de Leon, D.; Brin, M.F.; et al. The tor1a (dyt1) gene family and its role in early onset torsion dystonia. Genomics 1999, 62, 377–384.
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. Aaa+: A class of chaperone-like atpases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43.
- Rose, A.E.; Brown, R.S.; Schlieker, C. Torsins: Not your typical aaa+ atpases. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 532–549.
- Goodchild, R.E.; Dauer, W.T. The aaa+ protein torsina interacts with a conserved domain present in lap1 and a novel er protein. J. Cell Biol. 2005, 168, 855–862.
- Goodchild, R.E.; Dauer, W.T. Mislocalization to the nuclear envelope: An effect of the dystonia-causing torsina mutation. Proc. Natl. Acad. Sci. USA 2004, 101, 847–852.
- Zhu, L.; Wrabl, J.O.; Hayashi, A.P.; Rose, L.S.; Thomas, P.J. The torsin-family aaa+ protein ooc-5 contains a critical disulfide adjacent to sensor-ii that couples redox state to nucleotide binding. Mol. Biol. Cell 2008, 19, 3599–3612.
- Zhao, C.; Brown, R.S.; Chase, A.R.; Eisele, M.R.; Schlieker, C. Regulation of torsin atpases by lap1 and lull1. Proc. Natl. Acad. Sci. USA 2013, 110, E1545–E1554.
- Naismith, T.V.; Dalal, S.; Hanson, P.I. Interaction of torsina with its major binding partners is impaired by the dystonia-associated deltagag deletion. J. Biol. Chem. 2009, 284, 27866–27874.
- Brown, R.S.; Zhao, C.; Chase, A.R.; Wang, J.; Schlieker, C. The mechanism of torsin atpase activation. Proc. Natl. Acad. Sci. USA 2014, 111, E4822–E4831.
- Sosa, B.A.; Demircioglu, F.E.; Chen, J.Z.; Ingram, J.; Ploegh, H.; Schwartz, T.U. How lamina-associated polypeptide 1 (lap1) activates torsin. Elife 2014, 3, e03239.
- Demircioglu, F.E.; Sosa, B.A.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. Structures of torsina and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia. Elife 2016, 5, e17983.
- Zhu, L.; Millen, L.; Mendoza, J.L.; Thomas, P.J. A unique redox-sensing sensor ii motif in torsina plays a critical role in nucleotide and partner binding. J. Biol. Chem. 2010, 285, 37271–37280.
- Demircioglu, F.E.; Zheng, W.; McQuown, A.J.; Maier, N.K.; Watson, N.; Cheeseman, I.M.; Denic, V.; Egelman, E.H.; Schwartz, T.U. The aaa + atpase torsina polymerizes into hollow helical tubes with 8.5 subunits per turn. Nat. Commun. 2019, 10, 3262.
- Fichtman, B.; Zagairy, F.; Biran, N.; Barsheshet, Y.; Chervinsky, E.; Ben Neriah, Z.; Shaag, A.; Assa, M.; Elpeleg, O.; Harel, A.; et al. Combined loss of lap1b and lap1c results in an early onset multisystemic nuclear envelopathy. Nat. Commun. 2019, 10, 605.
- Dorboz, I.; Coutelier, M.; Bertrand, A.T.; Caberg, J.H.; Elmaleh-Berges, M.; Laine, J.; Stevanin, G.; Bonne, G.; Boespflug-Tanguy, O.; Servais, L. Severe dystonia, cerebellar atrophy, and cardiomyopathy likely caused by a missense mutation in tor1aip1. Orphanet J. Rare Dis. 2014, 9, 174.
- Kayman-Kurekci, G.; Talim, B.; Korkusuz, P.; Sayar, N.; Sarioglu, T.; Oncel, I.; Sharafi, P.; Gundesli, H.; Balci-Hayta, B.; Purali, N.; et al. Mutation in tor1aip1 encoding lap1b in a form of muscular dystrophy: A novel gene related to nuclear envelopathies. Neuromuscul. Disord. NMD 2014, 24, 624–633.
- Jungwirth, M.; Dear, M.L.; Brown, P.; Holbrook, K.; Goodchild, R. Relative tissue expression of homologous torsinb correlates with the neuronal specific importance of dyt1 dystonia-associated torsina. Hum. Mol. Genet. 2010, 19, 888–900.
- Kim, C.E.; Perez, A.; Perkins, G.; Ellisman, M.H.; Dauer, W.T. A molecular mechanism underlying the neural-specific defect in torsina mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9861–9866.
- Li, J.; Liang, C.-C.; Pappas, S.S.; Dauer, W.T. Torsinb overexpression prevents abnormal twisting in dyt1 dystonia mouse models. bioRxiv 2019, 836536.
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of aaa+ atpases. J. Struct. Biol. 2004, 146, 11–31.
- Hanson, P.I.; Whiteheart, S.W. Aaa+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529.
- Nagy, M.; Wu, H.C.; Liu, Z.; Kedzierska-Mieszkowska, S.; Zolkiewski, M. Walker-a threonine couples nucleotide occupancy with the chaperone activity of the aaa+ atpase clpb. Protein Sci. 2009, 18, 287–293.
- Scheffzek, K.; Ahmadian, M.R.; Wittinghofer, A. Gtpase-activating proteins: Helping hands to complement an active site. Trends Biochem. Sci. 1998, 23, 257–262.
- Schlieker, C.; Weibezahn, J.; Patzelt, H.; Tessarz, P.; Strub, C.; Zeth, K.; Erbse, A.; Schneider-Mergener, J.; Chin, J.W.; Schultz, P.G.; et al. Substrate recognition by the aaa+ chaperone clpb. Nat. Struct. Mol. Biol. 2004, 11, 607–615.
- Siddiqui, S.M.; Sauer, R.T.; Baker, T.A. Role of the processing pore of the clpx aaa+ atpase in the recognition and engagement of specific protein substrates. Genes Dev. 2004, 18, 369–374.
- Sauer, R.T.; Baker, T.A. Aaa+ proteases: Atp-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612.
- Goodchild, R.E.; Kim, C.E.; Dauer, W.T. Loss of the dystonia-associated protein torsina selectively disrupts the neuronal nuclear envelope. Neuron 2005, 48, 923–932.
- Liang, C.C.; Tanabe, L.M.; Jou, S.; Chi, F.; Dauer, W.T. Torsina hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J. Clin. Investig. 2014, 124, 3080–3092.
- Pappas, S.S.; Darr, K.; Holley, S.M.; Cepeda, C.; Mabrouk, O.S.; Wong, J.M.; LeWitt, T.M.; Paudel, R.; Houlden, H.; Kennedy, R.T.; et al. Forebrain deletion of the dystonia protein torsina causes dystonic-like movements and loss of striatal cholinergic neurons. Elife 2015, 4, e08352.
- DeSimone, J.C.; Pappas, S.S.; Febo, M.; Burciu, R.G.; Shukla, P.; Colon-Perez, L.M.; Dauer, W.T.; Vaillancourt, D.E. Forebrain knock-out of torsina reduces striatal free-water and impairs whole-brain functional connectivity in a symptomatic mouse model of dyt1 dystonia. Neurobiol. Dis. 2017, 106, 124–132.
- Hewett, J.W.; Kamm, C.; Boston, H.; Beauchamp, R.; Naismith, T.; Ozelius, L.; Hanson, P.I.; Breakefield, X.O.; Ramesh, V. Torsinb--perinuclear location and association with torsina. J. Neurochem. 2004, 89, 1186–1194.
- Jokhi, V.; Ashley, J.; Nunnari, J.; Noma, A.; Ito, N.; Wakabayashi-Ito, N.; Moore, M.J.; Budnik, V. Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep. 2013, 3, 988–995.
- Laudermilch, E.; Tsai, P.L.; Graham, M.; Turner, E.; Zhao, C.; Schlieker, C. Dissecting torsin/cofactor function at the nuclear envelope: A genetic study. Mol. Biol. Cell 2016, 27, 3964–3971.
- VanGompel, M.J.; Nguyen, K.C.; Hall, D.H.; Dauer, W.T.; Rose, L.S. A novel function for the caenorhabditis elegans torsin ooc-5 in nucleoporin localization and nuclear import. Mol. Biol. Cell 2015, 26, 1752–1763.
- Breakefield, X.O.; Kamm, C.; Hanson, P.I. Torsina: Movement at many levels. Neuron 2001, 31, 9–12.
- Fremont, R.; Tewari, A.; Angueyra, C.; Khodakhah, K. A role for cerebellum in the hereditary dystonia dyt1. Elife 2017, 6, e22775.
- Vander Heyden, A.B.; Naismith, T.V.; Snapp, E.L.; Hodzic, D.; Hanson, P.I. Lull1 retargets torsina to the nuclear envelope revealing an activity that is impaired by the dyt1 dystonia mutation. Mol. Biol. Cell 2009, 20, 2661–2672.
- Chase, A.R.; Laudermilch, E.; Wang, J.; Shigematsu, H.; Yokoyama, T.; Schlieker, C. Dynamic functional assembly of the torsin aaa+ atpase and its modulation by lap1. Mol. Biol. Cell 2017, 28, 2765–2772.
- Kariminejad, A.; Dahl-Halvarsson, M.; Ravenscroft, G.; Afroozan, F.; Keshavarz, E.; Goullee, H.; Davis, M.R.; Faraji Zonooz, M.; Najmabadi, H.; Laing, N.G.; et al. Tor1a variants cause a severe arthrogryposis with developmental delay, strabismus and tremor. Brain J. Neurol. 2017, 140, 2851–2859.
- Reichert, S.C.; Gonzalez-Alegre, P.; Scharer, G.H. Biallelic tor1a variants in an infant with severe arthrogryposis. Neurol. Genet. 2017, 3, e154.
- Isik, E.; Aykut, A.; Atik, T.; Cogulu, O.; Ozkinay, F. Biallelic tor1a mutations cause severe arthrogryposis: A case requiring reverse phenotyping. Eur. J. Med. Genet. 2019, 62, 103544.
- Gonzalez-Alegre, P. Advances in molecular and cell biology of dystonia: Focus on torsina. Neurobiol. Dis. 2019, 127, 233–241.
- Calakos, N.; Patel, V.D.; Gottron, M.; Wang, G.; Tran-Viet, K.N.; Brewington, D.; Beyer, J.L.; Steffens, D.C.; Krishnan, R.R.; Zuchner, S. Functional evidence implicating a novel tor1a mutation in idiopathic, late-onset focal dystonia. J. Med. Genet. 2010, 47, 646–650.
- Zirn, B.; Grundmann, K.; Huppke, P.; Puthenparampil, J.; Wolburg, H.; Riess, O.; Muller, U. Novel tor1a mutation p.Arg288gln in early-onset dystonia (dyt1). J. Neurol. Neurosurg. Psychiatry 2008, 79, 1327–1330.
- Leung, J.C.; Klein, C.; Friedman, J.; Vieregge, P.; Jacobs, H.; Doheny, D.; Kamm, C.; DeLeon, D.; Pramstaller, P.P.; Penney, J.B.; et al. Novel mutation in the tor1a (dyt1) gene in atypical early onset dystonia and polymorphisms in dystonia and early onset parkinsonism. Neurogenetics 2001, 3, 133–143.
- Cheng, F.B.; Feng, J.C.; Ma, L.Y.; Miao, J.; Ott, T.; Wan, X.H.; Grundmann, K. Combined occurrence of a novel tor1a and a thap1 mutation in primary dystonia. Mov. Disord. 2014, 29, 1079–1083.
- Vulinovic, F.; Lohmann, K.; Rakovic, A.; Capetian, P.; Alvarez-Fischer, D.; Schmidt, A.; Weissbach, A.; Erogullari, A.; Kaiser, F.J.; Wiegers, K.; et al. Unraveling cellular phenotypes of novel torsina/tor1a mutations. Hum. Mutat. 2014, 35, 1114–1122.
- Kock, N.; Naismith, T.V.; Boston, H.E.; Ozelius, L.J.; Corey, D.P.; Breakefield, X.O.; Hanson, P.I. Effects of genetic variations in the dystonia protein torsina: Identification of polymorphism at residue 216 as protein modifier. Hum. Mol. Genet. 2006, 15, 1355–1364.
- Dobricic, V.; Kresojevic, N.; Zarkovic, M.; Tomic, A.; Marjanovic, A.; Westenberger, A.; Cvetkovic, D.; Svetel, M.; Novakovic, I.; Kostic, V.S. Phenotype of non-c.907_909delgag mutations in tor1a: Dyt1 dystonia revisited. Parkinsonism Relat. Disord. 2015, 21, 1256–1259.
- Laudermilch, E.; Schlieker, C. Torsin atpases: Structural insights and functional perspectives. Curr. Opin. Cell Biol. 2016, 40, 1–7.
- Lessel, I.; Chen, M.J.; Luttgen, S.; Arndt, F.; Fuchs, S.; Meien, S.; Thiele, H.; Jones, J.R.; Shaw, B.R.; Crossman, D.K.; et al. Two novel cases further expand the phenotype of tor1aip1-associated nuclear envelopathies. Hum. Genet. 2020, 139, 483–498.
- Ghaoui, R.; Benavides, T.; Lek, M.; Waddell, L.B.; Kaur, S.; North, K.N.; MacArthur, D.G.; Clarke, N.F.; Cooper, S.T. Tor1aip1 as a cause of cardiac failure and recessive limb-girdle muscular dystrophy. Neuromuscul. Disord. NMD 2016, 26, 500–503.
- Risch, N.J.; Bressman, S.B.; Senthil, G.; Ozelius, L.J. Intragenic cis and trans modification of genetic susceptibility in dyt1 torsion dystonia. Am. J. Hum. Genet. 2007, 80, 1188–1193.
- Martino, D.; Gajos, A.; Gallo, V.; Cif, L.; Coubes, P.; Tinazzi, M.; Schneider, S.A.; Fiorio, M.; Zorzi, G.; Nardocci, N.; et al. Extragenetic factors and clinical penetrance of dyt1 dystonia: An exploratory study. J. Neurol. 2013, 260, 1081–1086.
- Santos, M.; Domingues, S.C.; Costa, P.; Muller, T.; Galozzi, S.; Marcus, K.; da Cruz e Silva, E.F.; da Cruz e Silva, O.A.; Rebelo, S. Identification of a novel human lap1 isoform that is regulated by protein phosphorylation. PLoS ONE 2014, 9, e113732.
- Rampello, A.J.; Laudermilch, E.; Vishnoi, N.; Prohet, S.M.; Shao, L.; Zhao, C.; Lusk, C.P.; Schlieker, C. Torsin atpases are required to complete nuclear pore complex biogenesis in interphase. bioRxiv 2019, 821835.
- Thaller, D.J.; Lusk, C.P. Fantastic nuclear envelope herniations and where to find them. Biochem. Soc. Trans. 2018, 46, 877–889.
- Pappas, S.S.; Liang, C.C.; Kim, S.; Rivera, C.O.; Dauer, W.T. Torsina dysfunction causes persistent neuronal nuclear pore defects. Hum. Mol. Genet. 2018, 27, 407–420.
- Olmos, Y.; Hodgson, L.; Mantell, J.; Verkade, P.; Carlton, J.G. Escrt-iii controls nuclear envelope reformation. Nature 2015, 522, 236–239.
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358.
- Gu, M.; LaJoie, D.; Chen, O.S.; von Appen, A.; Ladinsky, M.S.; Redd, M.J.; Nikolova, L.; Bjorkman, P.J.; Sundquist, W.I.; Ullman, K.S.; et al. Lem2 recruits chmp7 for escrt-mediated nuclear envelope closure in fission yeast and human cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2166–E2175.
- Webster, B.M.; Colombi, P.; Jager, J.; Lusk, C.P. Surveillance of nuclear pore complex assembly by escrt-iii/vps4. Cell 2014, 159, 388–401.
- Webster, B.M.; Thaller, D.J.; Jager, J.; Ochmann, S.E.; Borah, S.; Lusk, C.P. Chm7 and heh1 collaborate to link nuclear pore complex quality control with nuclear envelope sealing. EMBO J. 2016, 35, 2447–2467.
- Mackay, D.R.; Makise, M.; Ullman, K.S. Defects in nuclear pore assembly lead to activation of an aurora b-mediated abscission checkpoint. J. Cell Biol. 2010, 191, 923–931.
- Carlton, J.G.; Caballe, A.; Agromayor, M.; Kloc, M.; Martin-Serrano, J. Escrt-iii governs the aurora b-mediated abscission checkpoint through chmp4c. Science 2012, 336, 220–225.
- Morita, E.; Sandrin, V.; Chung, H.Y.; Morham, S.G.; Gygi, S.P.; Rodesch, C.K.; Sundquist, W.I. Human escrt and alix proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007, 26, 4215–4227.
- Rabut, G.; Lenart, P.; Ellenberg, J. Dynamics of nuclear pore complex organization through the cell cycle. Curr. Opin. Cell Biol. 2004, 16, 314–321.
- Chase, A.R.; Laudermilch, E.; Schlieker, C. Torsin atpases: Harnessing dynamic instability for function. Front. Mol. Biosci. 2017, 4, 29.

