Torsin ATPases are members of the AAA+ (ATPases associated with various cellular activities) superfamily of proteins, which participate in essential cellular processes. While AAA+ proteins are ubiquitously expressed and demonstrate distinct subcellular localizations, Torsins are the only AAA+ to reside within the nuclear envelope (NE) and endoplasmic reticulum (ER) network. Moreover, due to the absence of integral catalytic features, Torsins require the NE- and ER-specific regulatory cofactors, lamina-associated polypeptide 1 (LAP1) and luminal domain like LAP1 (LULL1), to efficiently trigger their atypical mode of ATP hydrolysis. Despite their implication in an ever-growing list of diverse processes, the specific contributions of Torsin/cofactor assemblies in maintaining normal cellular physiology remain largely enigmatic. Resolving gaps in the functional and mechanistic principles of Torsins and their cofactors are of considerable medical importance, as aberrant Torsin behavior is the principal cause of the movement disorder DYT1 early-onset dystonia.
- TorsinA
- nuclear pore complex (NPC)
- AAA+ ATPase
- lipin
- nuclear lamina
- nuclear envelopathy
- nuclear envelope
- muscular dystrophy
- myopathy
- cardiomyopathy
1. Introduction
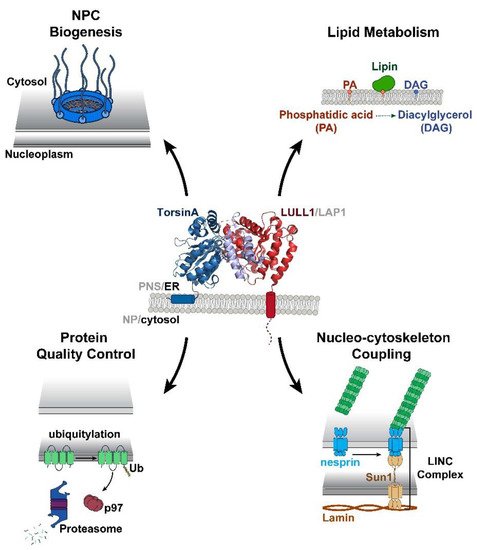
2. Structural and Biochemical Perspectives on Torsins
AAA+ ATPases are a class of P-loop NTPases with an evolutionarily conserved ATP-binding module that consists of two distinct subdomains: a large wedge-shaped N-terminal α/β RecA fold, and a small C-terminal α-helical domain (Figure 1). In addition to these defining tertiary structural features, all P-loop NTPases are further distinguished by a series of highly conserved amino acid sequence motifs, like the Walker motifs [3,38,39][3][21][22]. Torsins have historically been considered atypical or degenerate AAA+ proteins as they deviate from many of these canonical structural and functional features. Such variations between Torsins and their related ATPases, as well as across the Torsin family, might result from being the sole AAA+ ATPases to reside within the NE and ER, with the demands imposed by these subcellular locations molding their biological activities. Notably, the Walker A motif in Torsins (GXXXXGKN) diverges from the canonical sequence, and has been suggested to partially contribute to their reduced ATPase activity relative to other AAA+ proteins [8,40][8][23]. The most striking disparity between Torsins and other AAA+ ATPases, however, is their use of the NE- and ER-specific regulatory cofactors LAP1 and LULL1, respectively, to stimulate their unique mode of catalysis. Alone, Torsins lack the ability to hydrolyze ATP, a peculiarity that renders Torsin distinct from otherwise related Clp/Hsp100 proteins. An in vitro reconstitution of Torsin/cofactor assemblies made the Torsin system amenable to biochemical analysis, which demonstrated that cofactors are stimulators of Torsins’ ATPase activity [8]. Subsequent studies have since shown that ATP hydrolysis is achieved through an active site complementation mechanism in which either cofactor contributes a catalytic arginine residue reminiscent of an arginine finger [41][24], which is otherwise absent from all known members of the Torsin family [7,11,12,42][7][11][12][25]. Unlike its related Clp/Hsp100 chaperones, Torsins also lack conserved aromatic-hydrophobic pore loops [12,42][12][25] that commonly line the central channel of oligomeric AAA-ATPase ring assemblies and participate in substrate engagement and translocation [43,44][26][27]. This critical structural feature allows Clp/Hsp100 proteins to act as unfoldases or chaperones, or, when coupled to for example, protease modules, result in proteolysis of the substrate [39,45][22][28]. Although Torsins have been suggested to participate in numerous cellular activities including quality control and chaperone processes, the lack of pore loops and other AAA+ features present some uncertainty regarding their precise mechanistic roles to these pathways.3. Torsin Assemblies and Dystonia Movement Disorders
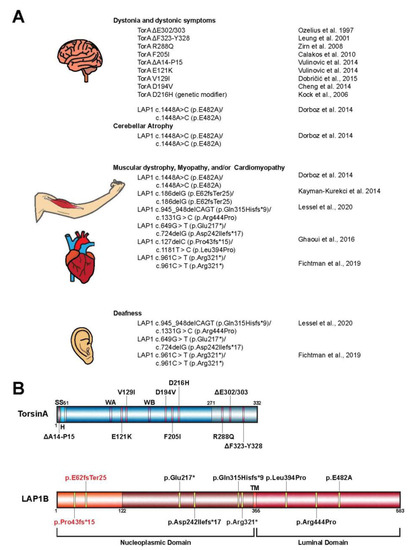
When considering the members of the Torsin family of ATPases, significant efforts have been placed in elucidating the biological roles of TorsinA due to its direct clinical significance in the highly debilitating movement disorder DYT1 early-onset dystonia [1]. Phenotypic effects of DYT1 dystonia usually manifest in early adolescence and are typified by sustained muscle contractions and involuntary twisting of individual or multiple muscle groups [1,15][1][15]. The most penetrant form of this disorder results from the autosomal dominant inheritance of a TOR1A variant bearing an in-frame deletion [1]. This mutation leads to the omission of a single glutamate residue in the C-terminal α-helical domain of the encoded protein, termed TorsinAΔE (Figure 2A,B) [12]. While the precise link between TorsinA dysfunction and DYT1 etiology remains unclear, considerable strides have been made over the past decade to further our understanding of the potential molecular and cellular pathways compromised in dystonia patients. Although Torsins are expressed in all mammalian tissues, TorsinA is distinctly enriched in a subset of cells in the nervous system in mice [18,46,47][18][29][30]. Thus, the differential expression pattern of Torsins likely contributes to the tissue-specific manifestation of TorsinAΔE-linked pathology. Indeed, phenotypes similar to DYT1 dystonia-specific symptoms were observed in conditional mouse models upon deletion of TorsinA in individual brain regions [47,48,49][30][31][32]. These symptoms manifest as abnormal posturing and dystonia-like twisting motions, as well as neurodegeneration of select regions of the central nervous system. The depletion of TorsinA also correlates with the appearance of aberrant protrusions of the INM into the perinuclear space [46,47][29][30]. This defect in NE architecture is observed upon manipulation of TorsinA in other model systems including human, fly, and worm cells, suggesting that the function of Torsins at the NE is evolutionarily conserved [27,30,50,51][33][34][35][36]. Thus, understanding the molecular and functional implications of Torsins in the contexts of these NE aberrations might therefore provide insight into the disease pathogenesis of DYT1 dystonia, as well as dynamic biological processes at the NE. From a genetic standpoint, the autosomal-dominant inheritance of DYT1 dystonia has long been considered to result from a dominant negative effect of the mutant TOR1A(ΔE) allele [52][37]. While the discussion of a loss-of-function vs. gain-of-function mechanism is still not fully resolved, a loss-of-function mechanism is consistent with the lack of cofactor-induced ATPase activity due to a failure of TorsinAΔE to productively interact with the cofactors LAP1/LULL1 [8,11,12][8][11][12]. In addition, TorsinA deletion or depletion causes dystonic symptoms even in animal models that do not express TorsinAΔE [47,49,53][30][32][38]. In fact, a mildly beneficial effect of expressing TorsinAΔE relative to a Torsin deletion was observed both in animal models [47,48][30][31] and tissue culture models [30][34], suggesting that TorsinAΔE may act as a hypomorphic allele. On the other hand, Torsins form dynamic higher-order oligomers [8,13,18,54][8][13][18][39] that rapidly disassemble upon cofactor binding and ATP hydrolysis [55][40]. Therefore, the addition of TorsinAΔE on the terminal position of TorsinA assemblies could interfere with the disassembly process thereby disrupting the equilibrium between Torsin oligomeric states and ultimately inhibiting their endogenous cellular activities. Moreover, the integration of TorsinAΔE into growing oligomers would represent a defective structure that could interfere with the formation of higher-order oligomers. However, all of these scenarios would require TorsinAΔE to bind to wild type TorsinA in a somewhat stable fashion, which is a questionable scenario given the dynamic instability of the Torsin system [55][40]. Accordingly, establishing the molecular interactions and functional mechanisms of Torsin/cofactor assemblies is critical for understanding DYT1 dystonia biology, as well as for the development of therapeutic strategies. Additional mutant alleles of TOR1A in patients with varying phenotypic severities have been reported (Figure 2A,B) [56,57,58,59,60,61,62,63,64,65,66][41][42][43][44][45][46][47][48][49][50][51]. Importantly, many of these mutations map to regions on TorsinA at the inter-subunit interface, suggesting they perturb Torsin/Torsin or Torsin/cofactor binding [4,12,67][4][12][52]. Supporting the idea that interrupting the Torsin/cofactor interaction is detrimental are reports of patients with mutations in the LAP1 gene, TOR1AIP1, who display dystonic-like symptoms [15[15][53],68], cardiomyopathy [14[14][15][53][54],15,68,69], deafness [14[14][53],68], and muscular dystrophy [16,69][16][54] (Figure 2A). Although many of these phenotypes arising from the TOR1AIP1 mutation are distinct from those observed in patients with TOR1A mutations, a subset of TOR1AIP1 patients experience dystonic symptoms similar to those presented by DYT1 dystonia (TOR1A mutation) patients. Along with the fact that no disease-causing mutations for LULL1 have been reported to date, these observations suggest that LAP1 and Torsins have independent molecular functions in addition to their obviously related contributions to cellular homeostasis. Though DYT1 dystonia results from the autosomal dominant inheritance of the TorsinAΔE-causing mutation, only ~30% of individuals with the mutant allele exhibit clinical features of the disease [70][55]. This reduced penetrance is difficult to rationalize and may suggest that additional biological or environmental risk factors contribute to the disease incidence [59,71][44][56].4. The Role of Torsin ATPases in Nuclear Pore Biogenesis
While Torsins display unique tissue-specific expression profiles and varying degrees of cofactor stimulation, their manipulation has been extensively linked to the formation of morphological abnormalities at the NE [27,30,46,50,51][33][34][29][35][36]. These structures, termed blebs, manifest as omega-shaped INM evaginations that project into the perinuclear space (Figure 3). Such malformations have been observed in numerous model organisms and cell lines, and are detected at early stages of embryonic development suggesting that Torsins perform a critical, evolutionarily conserved biological function at the NE. Recent studies utilizing a series of individual Torsin- and cofactor-deficient cell lines demonstrate that the genetic ablation of Torsins results in phenotypic traits reminiscent of those exhibited by primary mouse neurons harboring the DYT1-causing mutation [30,46][34][29]. A comparison of the blebbing phenotype further shows that blebs penetrant most in the altogether Torsin-deficient cell line compared to cell lines that lack only TorsinA and/or TorsinB [30][34]. This observation suggests that a functional redundancy exists between Torsin paralogs [30][34].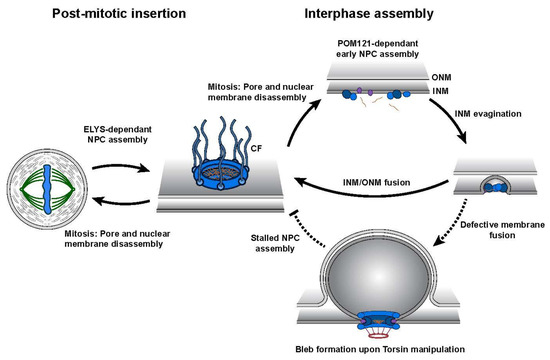
References
- Ozelius, L.J.; Hewett, J.W.; Page, C.E.; Bressman, S.B.; Kramer, P.L.; Shalish, C.; de Leon, D.; Brin, M.F.; Raymond, D.; Corey, D.P.; et al. The early-onset torsion dystonia gene (dyt1) encodes an atp-binding protein. Nat. Genet. 1997, 17, 40–48.
- Ozelius, L.J.; Page, C.E.; Klein, C.; Hewett, J.W.; Mineta, M.; Leung, J.; Shalish, C.; Bressman, S.B.; de Leon, D.; Brin, M.F.; et al. The tor1a (dyt1) gene family and its role in early onset torsion dystonia. Genomics 1999, 62, 377–384.
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. Aaa+: A class of chaperone-like atpases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43.
- Rose, A.E.; Brown, R.S.; Schlieker, C. Torsins: Not your typical aaa+ atpases. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 532–549.
- Goodchild, R.E.; Dauer, W.T. The aaa+ protein torsina interacts with a conserved domain present in lap1 and a novel er protein. J. Cell Biol. 2005, 168, 855–862.
- Goodchild, R.E.; Dauer, W.T. Mislocalization to the nuclear envelope: An effect of the dystonia-causing torsina mutation. Proc. Natl. Acad. Sci. USA 2004, 101, 847–852.
- Zhu, L.; Wrabl, J.O.; Hayashi, A.P.; Rose, L.S.; Thomas, P.J. The torsin-family aaa+ protein ooc-5 contains a critical disulfide adjacent to sensor-ii that couples redox state to nucleotide binding. Mol. Biol. Cell 2008, 19, 3599–3612.
- Zhao, C.; Brown, R.S.; Chase, A.R.; Eisele, M.R.; Schlieker, C. Regulation of torsin atpases by lap1 and lull1. Proc. Natl. Acad. Sci. USA 2013, 110, E1545–E1554.
- Naismith, T.V.; Dalal, S.; Hanson, P.I. Interaction of torsina with its major binding partners is impaired by the dystonia-associated deltagag deletion. J. Biol. Chem. 2009, 284, 27866–27874.
- Brown, R.S.; Zhao, C.; Chase, A.R.; Wang, J.; Schlieker, C. The mechanism of torsin atpase activation. Proc. Natl. Acad. Sci. USA 2014, 111, E4822–E4831.
- Sosa, B.A.; Demircioglu, F.E.; Chen, J.Z.; Ingram, J.; Ploegh, H.; Schwartz, T.U. How lamina-associated polypeptide 1 (lap1) activates torsin. Elife 2014, 3, e03239.
- Demircioglu, F.E.; Sosa, B.A.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. Structures of torsina and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia. Elife 2016, 5, e17983.
- Zhu, L.; Millen, L.; Mendoza, J.L.; Thomas, P.J. A unique redox-sensing sensor ii motif in torsina plays a critical role in nucleotide and partner binding. J. Biol. Chem. 2010, 285, 37271–37280.
- Demircioglu, F.E.; Zheng, W.; McQuown, A.J.; Maier, N.K.; Watson, N.; Cheeseman, I.M.; Denic, V.; Egelman, E.H.; Schwartz, T.U. The aaa + atpase torsina polymerizes into hollow helical tubes with 8.5 subunits per turn. Nat. Commun. 2019, 10, 3262.
- Fichtman, B.; Zagairy, F.; Biran, N.; Barsheshet, Y.; Chervinsky, E.; Ben Neriah, Z.; Shaag, A.; Assa, M.; Elpeleg, O.; Harel, A.; et al. Combined loss of lap1b and lap1c results in an early onset multisystemic nuclear envelopathy. Nat. Commun. 2019, 10, 605.
- Dorboz, I.; Coutelier, M.; Bertrand, A.T.; Caberg, J.H.; Elmaleh-Berges, M.; Laine, J.; Stevanin, G.; Bonne, G.; Boespflug-Tanguy, O.; Servais, L. Severe dystonia, cerebellar atrophy, and cardiomyopathy likely caused by a missense mutation in tor1aip1. Orphanet J. Rare Dis. 2014, 9, 174.
- Kayman-Kurekci, G.; Talim, B.; Korkusuz, P.; Sayar, N.; Sarioglu, T.; Oncel, I.; Sharafi, P.; Gundesli, H.; Balci-Hayta, B.; Purali, N.; et al. Mutation in tor1aip1 encoding lap1b in a form of muscular dystrophy: A novel gene related to nuclear envelopathies. Neuromuscul. Disord. NMD 2014, 24, 624–633.
- Jungwirth, M.; Dear, M.L.; Brown, P.; Holbrook, K.; Goodchild, R. Relative tissue expression of homologous torsinb correlates with the neuronal specific importance of dyt1 dystonia-associated torsina. Hum. Mol. Genet. 2010, 19, 888–900.
- Kim, C.E.; Perez, A.; Perkins, G.; Ellisman, M.H.; Dauer, W.T. A molecular mechanism underlying the neural-specific defect in torsina mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9861–9866.
- Li, J.; Liang, C.-C.; Pappas, S.S.; Dauer, W.T. Torsinb overexpression prevents abnormal twisting in dyt1 dystonia mouse models. bioRxiv 2019, 836536.
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of aaa+ atpases. J. Struct. Biol. 2004, 146, 11–31.
- Hanson, P.I.; Whiteheart, S.W. Aaa+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529.
- Nagy, M.; Wu, H.C.; Liu, Z.; Kedzierska-Mieszkowska, S.; Zolkiewski, M. Walker-a threonine couples nucleotide occupancy with the chaperone activity of the aaa+ atpase clpb. Protein Sci. 2009, 18, 287–293.
- Scheffzek, K.; Ahmadian, M.R.; Wittinghofer, A. Gtpase-activating proteins: Helping hands to complement an active site. Trends Biochem. Sci. 1998, 23, 257–262.
- Schlieker, C.; Weibezahn, J.; Patzelt, H.; Tessarz, P.; Strub, C.; Zeth, K.; Erbse, A.; Schneider-Mergener, J.; Chin, J.W.; Schultz, P.G.; et al. Substrate recognition by the aaa+ chaperone clpb. Nat. Struct. Mol. Biol. 2004, 11, 607–615.
- Siddiqui, S.M.; Sauer, R.T.; Baker, T.A. Role of the processing pore of the clpx aaa+ atpase in the recognition and engagement of specific protein substrates. Genes Dev. 2004, 18, 369–374.
- Sauer, R.T.; Baker, T.A. Aaa+ proteases: Atp-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612.
- Goodchild, R.E.; Kim, C.E.; Dauer, W.T. Loss of the dystonia-associated protein torsina selectively disrupts the neuronal nuclear envelope. Neuron 2005, 48, 923–932.
- Liang, C.C.; Tanabe, L.M.; Jou, S.; Chi, F.; Dauer, W.T. Torsina hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J. Clin. Investig. 2014, 124, 3080–3092.
- Pappas, S.S.; Darr, K.; Holley, S.M.; Cepeda, C.; Mabrouk, O.S.; Wong, J.M.; LeWitt, T.M.; Paudel, R.; Houlden, H.; Kennedy, R.T.; et al. Forebrain deletion of the dystonia protein torsina causes dystonic-like movements and loss of striatal cholinergic neurons. Elife 2015, 4, e08352.
- DeSimone, J.C.; Pappas, S.S.; Febo, M.; Burciu, R.G.; Shukla, P.; Colon-Perez, L.M.; Dauer, W.T.; Vaillancourt, D.E. Forebrain knock-out of torsina reduces striatal free-water and impairs whole-brain functional connectivity in a symptomatic mouse model of dyt1 dystonia. Neurobiol. Dis. 2017, 106, 124–132.
- Hewett, J.W.; Kamm, C.; Boston, H.; Beauchamp, R.; Naismith, T.; Ozelius, L.; Hanson, P.I.; Breakefield, X.O.; Ramesh, V. Torsinb--perinuclear location and association with torsina. J. Neurochem. 2004, 89, 1186–1194.
- Jokhi, V.; Ashley, J.; Nunnari, J.; Noma, A.; Ito, N.; Wakabayashi-Ito, N.; Moore, M.J.; Budnik, V. Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep. 2013, 3, 988–995.
- Laudermilch, E.; Tsai, P.L.; Graham, M.; Turner, E.; Zhao, C.; Schlieker, C. Dissecting torsin/cofactor function at the nuclear envelope: A genetic study. Mol. Biol. Cell 2016, 27, 3964–3971.
- VanGompel, M.J.; Nguyen, K.C.; Hall, D.H.; Dauer, W.T.; Rose, L.S. A novel function for the caenorhabditis elegans torsin ooc-5 in nucleoporin localization and nuclear import. Mol. Biol. Cell 2015, 26, 1752–1763.
- Breakefield, X.O.; Kamm, C.; Hanson, P.I. Torsina: Movement at many levels. Neuron 2001, 31, 9–12.
- Fremont, R.; Tewari, A.; Angueyra, C.; Khodakhah, K. A role for cerebellum in the hereditary dystonia dyt1. Elife 2017, 6, e22775.
- Vander Heyden, A.B.; Naismith, T.V.; Snapp, E.L.; Hodzic, D.; Hanson, P.I. Lull1 retargets torsina to the nuclear envelope revealing an activity that is impaired by the dyt1 dystonia mutation. Mol. Biol. Cell 2009, 20, 2661–2672.
- Chase, A.R.; Laudermilch, E.; Wang, J.; Shigematsu, H.; Yokoyama, T.; Schlieker, C. Dynamic functional assembly of the torsin aaa+ atpase and its modulation by lap1. Mol. Biol. Cell 2017, 28, 2765–2772.
- Kariminejad, A.; Dahl-Halvarsson, M.; Ravenscroft, G.; Afroozan, F.; Keshavarz, E.; Goullee, H.; Davis, M.R.; Faraji Zonooz, M.; Najmabadi, H.; Laing, N.G.; et al. Tor1a variants cause a severe arthrogryposis with developmental delay, strabismus and tremor. Brain J. Neurol. 2017, 140, 2851–2859.
- Reichert, S.C.; Gonzalez-Alegre, P.; Scharer, G.H. Biallelic tor1a variants in an infant with severe arthrogryposis. Neurol. Genet. 2017, 3, e154.
- Isik, E.; Aykut, A.; Atik, T.; Cogulu, O.; Ozkinay, F. Biallelic tor1a mutations cause severe arthrogryposis: A case requiring reverse phenotyping. Eur. J. Med. Genet. 2019, 62, 103544.
- Gonzalez-Alegre, P. Advances in molecular and cell biology of dystonia: Focus on torsina. Neurobiol. Dis. 2019, 127, 233–241.
- Calakos, N.; Patel, V.D.; Gottron, M.; Wang, G.; Tran-Viet, K.N.; Brewington, D.; Beyer, J.L.; Steffens, D.C.; Krishnan, R.R.; Zuchner, S. Functional evidence implicating a novel tor1a mutation in idiopathic, late-onset focal dystonia. J. Med. Genet. 2010, 47, 646–650.
- Zirn, B.; Grundmann, K.; Huppke, P.; Puthenparampil, J.; Wolburg, H.; Riess, O.; Muller, U. Novel tor1a mutation p.Arg288gln in early-onset dystonia (dyt1). J. Neurol. Neurosurg. Psychiatry 2008, 79, 1327–1330.
- Leung, J.C.; Klein, C.; Friedman, J.; Vieregge, P.; Jacobs, H.; Doheny, D.; Kamm, C.; DeLeon, D.; Pramstaller, P.P.; Penney, J.B.; et al. Novel mutation in the tor1a (dyt1) gene in atypical early onset dystonia and polymorphisms in dystonia and early onset parkinsonism. Neurogenetics 2001, 3, 133–143.
- Cheng, F.B.; Feng, J.C.; Ma, L.Y.; Miao, J.; Ott, T.; Wan, X.H.; Grundmann, K. Combined occurrence of a novel tor1a and a thap1 mutation in primary dystonia. Mov. Disord. 2014, 29, 1079–1083.
- Vulinovic, F.; Lohmann, K.; Rakovic, A.; Capetian, P.; Alvarez-Fischer, D.; Schmidt, A.; Weissbach, A.; Erogullari, A.; Kaiser, F.J.; Wiegers, K.; et al. Unraveling cellular phenotypes of novel torsina/tor1a mutations. Hum. Mutat. 2014, 35, 1114–1122.
- Kock, N.; Naismith, T.V.; Boston, H.E.; Ozelius, L.J.; Corey, D.P.; Breakefield, X.O.; Hanson, P.I. Effects of genetic variations in the dystonia protein torsina: Identification of polymorphism at residue 216 as protein modifier. Hum. Mol. Genet. 2006, 15, 1355–1364.
- Dobricic, V.; Kresojevic, N.; Zarkovic, M.; Tomic, A.; Marjanovic, A.; Westenberger, A.; Cvetkovic, D.; Svetel, M.; Novakovic, I.; Kostic, V.S. Phenotype of non-c.907_909delgag mutations in tor1a: Dyt1 dystonia revisited. Parkinsonism Relat. Disord. 2015, 21, 1256–1259.
- Laudermilch, E.; Schlieker, C. Torsin atpases: Structural insights and functional perspectives. Curr. Opin. Cell Biol. 2016, 40, 1–7.
- Lessel, I.; Chen, M.J.; Luttgen, S.; Arndt, F.; Fuchs, S.; Meien, S.; Thiele, H.; Jones, J.R.; Shaw, B.R.; Crossman, D.K.; et al. Two novel cases further expand the phenotype of tor1aip1-associated nuclear envelopathies. Hum. Genet. 2020, 139, 483–498.
- Ghaoui, R.; Benavides, T.; Lek, M.; Waddell, L.B.; Kaur, S.; North, K.N.; MacArthur, D.G.; Clarke, N.F.; Cooper, S.T. Tor1aip1 as a cause of cardiac failure and recessive limb-girdle muscular dystrophy. Neuromuscul. Disord. NMD 2016, 26, 500–503.
- Risch, N.J.; Bressman, S.B.; Senthil, G.; Ozelius, L.J. Intragenic cis and trans modification of genetic susceptibility in dyt1 torsion dystonia. Am. J. Hum. Genet. 2007, 80, 1188–1193.
- Martino, D.; Gajos, A.; Gallo, V.; Cif, L.; Coubes, P.; Tinazzi, M.; Schneider, S.A.; Fiorio, M.; Zorzi, G.; Nardocci, N.; et al. Extragenetic factors and clinical penetrance of dyt1 dystonia: An exploratory study. J. Neurol. 2013, 260, 1081–1086.
- Santos, M.; Domingues, S.C.; Costa, P.; Muller, T.; Galozzi, S.; Marcus, K.; da Cruz e Silva, E.F.; da Cruz e Silva, O.A.; Rebelo, S. Identification of a novel human lap1 isoform that is regulated by protein phosphorylation. PLoS ONE 2014, 9, e113732.
- Rampello, A.J.; Laudermilch, E.; Vishnoi, N.; Prohet, S.M.; Shao, L.; Zhao, C.; Lusk, C.P.; Schlieker, C. Torsin atpases are required to complete nuclear pore complex biogenesis in interphase. bioRxiv 2019, 821835.
- Thaller, D.J.; Lusk, C.P. Fantastic nuclear envelope herniations and where to find them. Biochem. Soc. Trans. 2018, 46, 877–889.
- Pappas, S.S.; Liang, C.C.; Kim, S.; Rivera, C.O.; Dauer, W.T. Torsina dysfunction causes persistent neuronal nuclear pore defects. Hum. Mol. Genet. 2018, 27, 407–420.
- Olmos, Y.; Hodgson, L.; Mantell, J.; Verkade, P.; Carlton, J.G. Escrt-iii controls nuclear envelope reformation. Nature 2015, 522, 236–239.
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358.
- Gu, M.; LaJoie, D.; Chen, O.S.; von Appen, A.; Ladinsky, M.S.; Redd, M.J.; Nikolova, L.; Bjorkman, P.J.; Sundquist, W.I.; Ullman, K.S.; et al. Lem2 recruits chmp7 for escrt-mediated nuclear envelope closure in fission yeast and human cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2166–E2175.
- Webster, B.M.; Colombi, P.; Jager, J.; Lusk, C.P. Surveillance of nuclear pore complex assembly by escrt-iii/vps4. Cell 2014, 159, 388–401.
- Webster, B.M.; Thaller, D.J.; Jager, J.; Ochmann, S.E.; Borah, S.; Lusk, C.P. Chm7 and heh1 collaborate to link nuclear pore complex quality control with nuclear envelope sealing. EMBO J. 2016, 35, 2447–2467.
- Mackay, D.R.; Makise, M.; Ullman, K.S. Defects in nuclear pore assembly lead to activation of an aurora b-mediated abscission checkpoint. J. Cell Biol. 2010, 191, 923–931.
- Carlton, J.G.; Caballe, A.; Agromayor, M.; Kloc, M.; Martin-Serrano, J. Escrt-iii governs the aurora b-mediated abscission checkpoint through chmp4c. Science 2012, 336, 220–225.
- Morita, E.; Sandrin, V.; Chung, H.Y.; Morham, S.G.; Gygi, S.P.; Rodesch, C.K.; Sundquist, W.I. Human escrt and alix proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007, 26, 4215–4227.
- Rabut, G.; Lenart, P.; Ellenberg, J. Dynamics of nuclear pore complex organization through the cell cycle. Curr. Opin. Cell Biol. 2004, 16, 314–321.
- Chase, A.R.; Laudermilch, E.; Schlieker, C. Torsin atpases: Harnessing dynamic instability for function. Front. Mol. Biosci. 2017, 4, 29.