 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Martin Holcik | + 2610 word(s) | 2610 | 2020-06-02 08:14:27 | | | |
| 2 | Nicole Yin | -52 word(s) | 2558 | 2020-11-05 11:00:08 | | |
Video Upload Options
tRNA nucleotidyl transferase 1 (TRNT1) is an essential enzyme catalyzing the addition of terminal cytosine-cytosine-adenosine (CCA) trinucleotides to all mature tRNAs, which is necessary for aminoacylation. It was recently discovered that partial loss-of-function mutations in TRNT1 are associated with various, seemingly unrelated human diseases including sideroblastic anemia with B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD), retinitis pigmentosa with erythrocyte microcytosis, and progressive B-cell immunodeficiency. In addition, even within the same disease, the severity and range of the symptoms vary greatly, suggesting a broad, pleiotropic impact of imparting TRNT1 function on diverse cellular systems. This entry describes the current state of knowledge of the TRNT1 function and the phenotypes associated with mutations in TRNT1.
1. TRNT1 Overview
Maturation of tRNAs (both mitochondrial and cytoplasmic) is a multi-step process that requires 5′ and 3′ processing, extensive base modifications, CCA addition, and aminoacylation. For cytoplasmic tRNAs, these steps occur in a precise order and are performed in distinct cellular compartments; in contrast, all mt tRNA processing steps occur in mitochondria [1]. The CCA addition is performed by a unique CCA-adding enzyme, tRNA nucleotidyltransferase (TRNT1 in human, or CCA1 in yeast; EC 2.7.7.72), which is located on the third human chromosome at position 26.2 and has 7 exons with a span of approximately 20kb [2]. TRNT1 encodes the only human CCA-adding enzyme, an RNA polymerase required for the post-transcriptional, template-independent addition of two cytosines and one adenosine to the 3′ end of both cytosolic and mitochondrial tRNAs [2]. This 3′ addition is required for aminoacylation, correct positioning on the ribosome, and subsequent protein synthesis [3]. Following the addition, the CCA trinucleotide acts as an anti-determinant for 3′ endoribonuclease activity [4]. In the mitochondria, the addition of CCA (and subsequent 3′ protection) has been proposed to stabilize select tRNA in order to achieve the balanced tRNA levels observed in the mitochondria [5]. The 3′ CCA tRNA terminus is subject to frequent cleavage or degradation and TRNT1 also functions to maintain and repair previously added CCA sequence [6]. Importantly, TRNT1 can discriminate against tRNA backbone damage, keeping damaged tRNAs from incorporation into the translation machinery (see below) [7]. Of note, TRNT1 appears to play additional, non-canonical roles in the cell. These include assisting in tRNA nuclear export and quality control as a nuclear export adaptor protein [8], the addition of CCA to small RNAs [9][10], and assessment of tRNA structure and targeting of unstable tRNA for decay which will be briefly described within this entry [11][9].
TRNT1 is classified in full as a template-independent class II RNA nucleotidyltransferase enzyme. Although similar in function to each other, the class II CCA-adding enzymes differ in sequence significantly from the class I enzymes. In addition, class I and class II enzymes are found in different life kingdoms. Class I are present in archaeal organisms, while class II are found in eukaryotes [12]. Although the sequence differences inform the specific mechanism of CCA addition, there is quite a bit of homology between the active sites of class I and class II enzymes hinting at common basic principles of CCA addition. Furthermore, it has been found that in some bacteria, the CCA addition is carried out by two separate enzymes—one which is responsible for adding “CC” and another one for adding an “A”. The flexibility between the head and neck domain of the CCA-adding enzyme is what allows for the CCA trinucleotide addition to be completed by a single enzyme (see below) [12]. Class I and class II CCA-adding enzymes share similarities to other polymerase enzymes, specifically within their active sites. However, CCA-adding enzymes are unique in that they are template-independent polymerases. This means that they do not add the CCA end to tRNA based off a pre-existing sequence encoded by the genome, rather the 3D structure of the enzyme and its amino acid sequence itself select for the specific nucleotide bases (C, C, or A) to be added in a specific and precise sequence [3]. For this entry, we will only be focusing on TRNT1, the full 3D structure of which can be seen in Figure 1. It should be noted, however, that the specific mechanistic details of class I enzymes function are more robustly understood than those of their eukaryotic counterparts [12].
2. TRNT1 Structure and Mechanism of Action
When it comes to the structure of TRNT1, the protein can be divided into four distinct functional domains: head, neck, body, and tail (Figure 1, Figure 2) [12]. Each of these domains fulfills a specific function within TRNT1; however, the two most critical domains are the head and neck, as these are the domains that house and assist in the movement of the active site for CCA addition. The enzyme active site is comprised of two aspartic acid residues, Asp48 and Asp50, and is located within the head domain [7]. There are two other important residues in the head domain—glutamic acid, Glu164, and arginine, Arg168—which, due to the flexible properties of their side chains, allows the binding pocket of the active site to discriminate between cytosines and adenines, and incorporate the correct nucleotide at each step of CCA addition. The second most important domain of TRNT1 protein is the neck domain, which interacts with the head domain to give the enzyme its flexibility. Between the head and neck domains, there is a positively charged region of residues where the tRNA itself binds to TRNT1. The 5′ end of the acceptor stem is the part of the tRNA which is tightly bound to TRNT1, while the 3′ end is more loosely attached [13]. This interesting aspect of the RNA–protein complex formation is what allows for the tRNA to be tightly held to TRNT1, while at the same time allowing the 3′ end to move and be freely added to. The body and tail regions of the TRNT1 protein are not important for the CCA-adding function of the active site but are responsible for identifying the structural components which define the tRNA, such as acceptor stem structure [12].
The mechanistic steps of CCA addition by TRNT1 are briefly outlined in Figure 2A. When TRNT1 encounters a tRNA molecule lacking a CCA 3′ end, or with a damaged 3′ end the enzyme will form an RNA–protein complex with the tRNA, binding tightly to the 5′ end of the acceptor stem [12]. Through this attachment, TRNT1 adopts its first conformational state, which prompts the addition of the first cytosine to the 3′ end of the tRNA. The addition of the first C to the 3′ end causes the head-neck joint to flex and positions the active site into a conformation which favors the addition of the second C. After both cytosines have been added, the RNA–protein complex adopts its final conformation, which allows for the adenosine to be added. With the complete CCA terminus now constructed, the RNA–protein complex dissociates and the tRNA is released. The release of tRNA allows for TRNT1 to return to its original conformation, and to engage another tRNA molecule. It should be noted that this is the current model for CCA addition by class II CCA-adding enzymes; however, the precise molecular steps are still not fully understood.
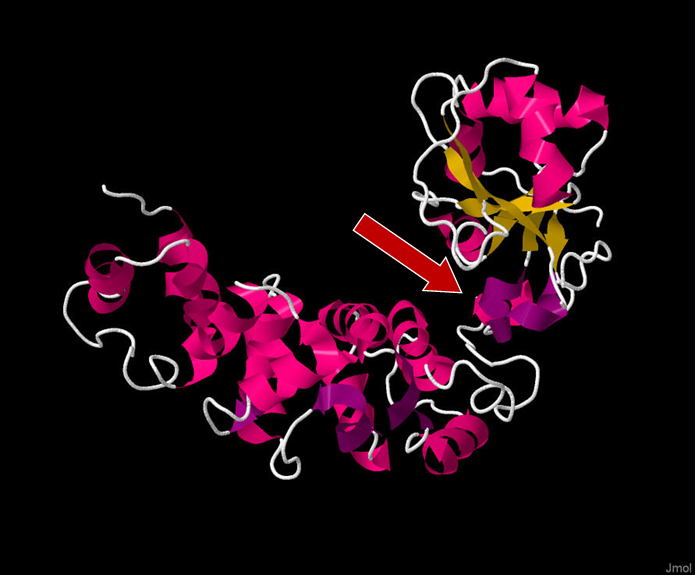
Figure 1. Full 3D structure of TRNT1 (tRNA Nucleotidyl Transferase 1), with the head domain being on the top right side of the figure. The red arrow indicates the positively charged cleft between the head and neck domain which acts as the active site of the enzyme. Viewed in Jmol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org/. Sequence taken from http://www.rcsb.org/structure/1ou5
3. TRNT1 and tRNA Quality Control
Another important function of the TRNT1 protein is its role in tRNA quality control through the addition of secondary CCA ends as well as the repair of damaged CCA ends. All organisms possess a functional homolog of TRNT1, be it a class I or class II tRNA nucleotidyltransferase enzyme. Interestingly, however, not all organisms require a post-transcriptional CCA-adding function. For example, in Escherichia coli, the 3′ CCA end is already encoded within the DNA genes for tRNAs, and the correct terminal CCA is added during transcription [7]. Yet, E. coli still possesses a CCA-adding enzyme [7]. It is believed that the tRNA nucleotidyltransferase enzyme is still required due to its ability to recognize and repair CCA ends, which are vulnerable to damage during the tRNAs life [7]. The 3′ CCA ends of all tRNAs are essential for the proper function of the aminoacyl-tRNA synthetases (aaRS), a class of enzymes that are responsible for the addition of the amino acids to their cognate tRNAs [12]. The damage to CCA termini can result in improper aminoacylation, where the incorrect amino acids could be attached to the wrong tRNAs [13]. Consequently, such mis-aminoacylation could result in mutations in proteins at the level of translation due to incorrect amino acids being incorporated into the proteins. TRNT1 can detect and repair partially damaged CCA termini [12].
Figure 2B outlines an additional mechanism of tRNA quality control performed by TRNT1, one that detects structurally unstable tRNAs and tags them for degradation. The structural quality of tRNAs is paramount for their ability to deliver appropriate amino acids to the translation apparatus. Unstable tRNAs may arise due to loss of base modifications that stabilize secondary tRNA structure, or due to point mutations disrupting base pairing [14]. During the CCA addition cycle to a tRNA, the TRNT1 enzyme exerts pressure onto the tRNA, which creates tension in the tRNA. If the tRNA is structurally sound, then this tension does not affect the structure of the tRNA, the RNA–protein complex dissociates properly to release the modified tRNA, and TRNT1 resets to its natural position ready to engage another tRNA [13]. However, if the tRNA is structurally unstable, it will “buckle” under the pressure and will undergo additional modification by TRNT1. Under the pressure of TRNT1, the tRNA will “slip” backward on its 3′ end and form a temporary bubble upstream of the 3′ end. This results in the CC of the CCA end binding with their cognate nucleotides (G) on the 5′ end. This slippage and formation of the temporary nucleotide bubble allow the tRNA to appear to TRNT1 as if it does not currently have a CCA end attached. Due to this, TRNT1 will revert to its default conformation and begins the second round of CCA addition. On the completion of this second CCA addition, the RNA–protein complex is dissociated and the released tRNA is stretched back out so that the CCA end is free and not bound to any nucleotides on the 5′ end of the tRNA. This newly formed CCACCA sequence functions as a signal to 3′–5′ exonucleases that target the tRNA for degradation [13]. Therefore, through the process of the second CCA addition TRNT1 can identify structurally unstable tRNAs, and mark them for degradation to prevent these aberrant molecules from potentially negatively impacting protein synthesis. It is important to note that TRNT1 can only recognize and tag for degradation structurally unstable tRNA that contains GG sequence at the 5′ end of the acceptor stem, which is present in approximately 45% of all tRNAs [11]. For all other tRNAs, a CCACCA-independent mechanism executed by poly(A) polymerases is used [12].
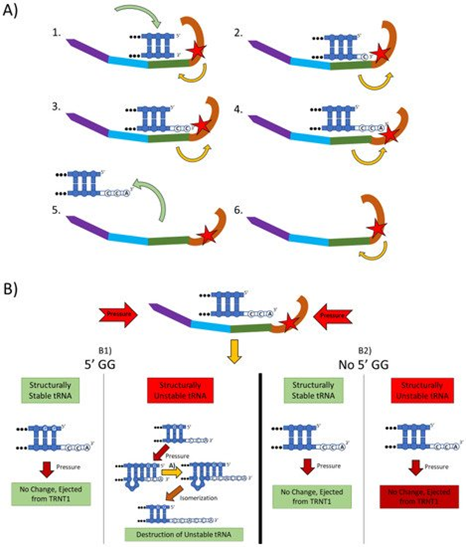
Figure 2. The two main functions of the TRNT1 enzyme: CCA addition, and tRNA quality control through CCA addition. (A) CCA addition. TRNT1 enzyme pictured with domains colored as: head (orange), neck (green), body (blue), and tail (purple). Additionally, the catalytic site is visualized by the red star, with the white nucleotides representing the end of the acceptor stem of the attached tRNA, and the yellow arrows indicating movement between the head and neck domains. (A1) tRNA binds with TRNT1 forming an RNA–protein complex. (A2) Cytosine is incorporated in a template-independent manner to the 3′ end of the tRNA, resulting in a conformational change in the TRNT1 enzyme. (A3) A second cytosine is added to the growing 3′ end of the tRNA, resulting in an additional structural shift in TRNT1. (A4) The terminal adenine is added to finish the CCA addition, and the structural integrity of the tRNA is judged. (A5) The RNA–protein complex dissociates and the tRNA is ejected from TRNT1. (A6) TRNT1 resets to its resting conformation. (B) tRNA quality control. During the addition of the CCA end to the tRNA, the TRNT1 exerts pressure on the tRNA, which can result in one of four outcomes. (B1) If the tRNA has two guanines on its 5′ end and the tRNA is structurally unstable, the pressure from TRNT1 will force the recently created CCA end to slide back and create a bubble of nucleotides. This allows for the addition of a second CCA end, tagging the tRNA for destruction. If the two 5′ guanines are present and the tRNA is structurally sound, then no bubble will form and no second CCA will be added. (B2) If there is no GG present on the 5-prime end of the tRNA, when the pressure is exerted from the TRNT1, no bubble is formed and the tRNA is ejected from the RNA–protein complex. This will occur whether the tRNA is structurally stable or not.
4. TRNT1 and Disease
Although CCA addition has been studied extensively in prokaryotes and lower eukaryotes for over thirty years [15], the connection between TRNT1 and human disease has been made only recently. Early genetic mapping of the candidate genes associated with non-syndromic mental retardation on chromosome3p (MRT2A) identified a region containing nine genes including TRNT1[16]. Although all the affected individuals contained a homozygous G→A base pair change in a 30–splice site of TRNT1, 30% of the normal controls also harboured this mutation. TRNT1was also identified in a differential gene expression study comparing colorectal cancer patients of African American and European origins [17]. The TRNT1 was shown to be downregulated in the cohort of African American patients, suggesting a link with higher incidence and mortality of colorectal cancer seen in African Americans. However, the first direct link between TRNT1 and disease was reported only in 2014. At that time, a young female patient presented with sensorineural hearing impairment, sideroblastic anemia, hyperalaninemia, episodic fever, B-cell immunodeficiency, and developmental delay (SIFD; OMIM #616084). SIFD is a recently identified severe syndromic form of congenital sideroblastic anemia [18]. A whole-exome sequencing strategy was therefore pursued to identify disease-causing mutations in this patient. Two non-conservative missense mutations in phylogenetically highly conserved residues were found in theTRNT1. In parallel, hypomorphic mutations in the same gene were identified in a cohort of additional 10 clinically similar patients using homozygosity mapping in nine pedigrees. Using biochemical, cell, and yeast approaches, it was demonstrated that the patients’ mutations are indeed causative of SIFD [19]. This conclusion was subsequently confirmed by an independent group that additionally showed defective CCA addition to the non-canonical mitochondrial tRNASer(AGY), but not other mitochondrial or cytoplasmic tRNAs, and reduced mitochondrial translation in patient samples [20]. In contrast, a similar investigation of an independent patient by Wedatilake et al. [21] disclosed a partial lack of CCA in mt-tRNACys and mt-tRNALeu(UUR), but not in tRNASer(AGY). Others have further shown that although patient fibroblasts do not appear to be defective in cellular morphology, TRNT1 localization, global translation, mitochondrial network architecture, or mitochondrial transmembrane potential the TRNT1 patient-derived fibroblast cells are defective in their ability to properly form OXPHOS complexes and consume oxygen, defects that likely contribute to the observed mitochondrial phenotypes associated with mutant TRNT1 [22]. SIFD manifests with a wide range of possible symptoms that continue to grow as new cases of SIFD are identified, each with different causative mutations within the TRNT1 gene (See paper linked below for the full list of symptoms).
References
- Anita K. Hopper; tRNA transfers to the limelight. Genes & Development 2003, 17, 162-180, 10.1101/gad.1049103.
- Takashi Nagaike; Tsutomu Suzuki; Yukihide Tomari; Chie Takemoto-Hori; Fumiko Negayama; Kimitsuna Watanabe; Takuya Ueda; Identification and Characterization of Mammalian Mitochondrial tRNA nucleotidyltransferases. Journal of Biological Chemistry 2001, 276, 40041-40049, 10.1074/jbc.m106202200.
- Shi, P.Y.; CCA addition by tRNA nucleotidyltransferase: polymerization without translocation?. EMBO J 1998, 17, 3197.
- Amulya Mohan; Stacey Whyte; Xudong Wang; Masayuki Nashimoto; Louis Levinger; The 3′ end CCA of mature tRNA is an antideterminant for eukaryotic 3′-tRNase. RNA 1999, 5, 245-256, 10.1017/s1355838299981256.
- Mohan, A.; Human mitochondrial tRNA quality control in health and disease: a channelling mechanism?. RNA Biology 1999, 5, 245.
- Ya-Ming Hou; CCA addition to tRNA: Implications for tRNA quality control. IUBMB Life 2010, 62, 251-60, 10.1002/iub.301.
- Martin A Augustin; Andreas S. Reichert; Heike Betat; Robert Huber; Mario Moerl; Clemens Steegborn; Crystal Structure of the Human CCA-adding Enzyme: Insights into Template-independent Polymerization. Journal of Molecular Biology 2003, 328, 985-994, 10.1016/s0022-2836(03)00381-4.
- Feng, W.; A Los1p-independent pathway for nuclear export of intronless tRNAs in Saccharomycescerevisiae.. Proc Natl Acad Sci U S A 2002, 99, 5412.
- Claus-D. Kuhn; Jeremy E. Wilusz; Yuxuan Zheng; Peter A. Beal; Leemor Joshua-Tor; On-enzyme refolding permits small RNA and tRNA surveillance by the CCA-adding enzyme.. Cell 2015, 160, 644-658, 10.1016/j.cell.2015.01.005.
- HyunDae D. Cho; Kozo Tomita; Tsutomu Suzuki; Alan M. Weiner; U2 Small Nuclear RNA Is a Substrate for the CCA-adding Enzyme (tRNA Nucleotidyltransferase). Journal of Biological Chemistry 2001, 277, 3447-3455, 10.1074/jbc.m109559200.
- Wilusz, J.E.; tRNAs marked with CCACCA are targeted for degradation. Science 2011, 334, 817.
- Kozo Tomita; Seisuke Yamashita; Molecular mechanisms of template-independent RNA polymerization by tRNA nucleotidyltransferases. Frontiers in Genetics 2014, 5, 36, 10.3389/fgene.2014.00036.
- Heike Betat; Mario Moerl; The CCA-adding enzyme: A central scrutinizer in tRNA quality control. BioEssays 2015, 37, 975-982, 10.1002/bies.201500043.
- Christian Lorenz; Christina E. Lünse; Mario Moerl; tRNA Modifications: Impact on Structure and Thermal Adaptation. Biomolecules 2017, 7, 35, 10.3390/biom7020035.
- Mathias Sprinzl; Friedrich Cramer; The -C-C-A End of tRNA and Its Role in Protein Biosynthesis. Progress in Nucleic Acid Research and Molecular Biology Volume 74 1978, 22, 1-69, 10.1016/s0079-6603(08)60798-9.
- Joseph J. Higgins; J Pucilowska; Rq Lombardi; John Rooney; Candidate genes for recessive non-syndromic mental retardation on chromosome 3p (MRT2A)*. Clinical Genetics 2004, 65, 496-500, 10.1111/j.0009-9163.2004.00267.x.
- Jovov, B.; Differential gene expression between African American and European American colorectal cancer patients. PLoS One 2012, 7, e30168.
- Daniel Wiseman; Alison May; Stephen Jolles; Philip Connor; Colin Powell; Matthew Heeney; Patricia J. Giardina; Robert J. Klaassen; Pranesh Chakraborty; Michael T. Geraghty; et al.Nathalie Major-CookCaroline KannengiesserIsabelle ThuretAlexis A. ThompsonLaura MarquesStephen HughesDenise K. BonneySylvia S. BottomleyMark D. FlemingRobert F. K. Wynn A novel syndrome of congenital sideroblastic anemia, B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD). Blood 2013, 122, 112-123, 10.1182/blood-2012-08-439083.
- Chakraborty, P.; Mutations in TRNT1, encoding the CCA-adding enzyme, cause congenital sideroblastic anemia with B cell immunodeficiency, periodic fevers and developmental delay (SIFD). Blood 2014, 5, 2014.
- Florin Sasarman; Isabelle Thiffault; Woranontee Weraarpachai; Steven Salomon; Catalina Maftei; Julie Gauthier; Benjamin Ellazam; Neil Webb; Hana Antonicka; Alexandre Janer; et al.Catherine Brunel-GuittonOrly ElpelegGrant MitchellEric A Shoubridge The 3′ addition of CCA to mitochondrial tRNASer(AGY) is specifically impaired in patients with mutations in the tRNA nucleotidyl transferase TRNT1. Human Molecular Genetics 2015, 24, 2841-2847, 10.1093/hmg/ddv044.
- Yehani Wedatilake; Rojeen Niazi; Elisa Fassone; Christopher Powell; Sarah Pearce; Vincent Plagnol; José W. Saldanha; Robert Kleta; W Kling Chong; Emma Footitt; et al.Philippa MillsJan-Willem TaanmanMichal MinczukPeter T. ClaytonShamima Rahman TRNT1 deficiency: clinical, biochemical and molecular genetic features. Orphanet Journal of Rare Diseases 2016, 11, 90, 10.1186/s13023-016-0477-0.
- Urszula Liwak-Muir; Hapsatou Mamady; Thierry Naas; Quinlan Wylie; Skye McBride; Matthew Lines; Jean Michaud; Stephen D. Baird; Pranesh Chakraborty; Martin Holcik; et al. Impaired activity of CCA-adding enzyme TRNT1 impacts OXPHOS complexes and cellular respiration in SIFD patient-derived fibroblasts.. Orphanet Journal of Rare Diseases 2016, 11, 79, 10.1186/s13023-016-0466-3.

