 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Abed Agbarya | + 2299 word(s) | 2299 | 2021-06-23 11:09:38 | | | |
| 2 | Bruce Ren | -21 word(s) | 2278 | 2021-06-29 10:29:43 | | |
Video Upload Options
The association between cancer and thrombosis has been known for over a century and a half. However, the mechanisms that underlie this correlation are not fully characterized. Hypercoagulability in cancer patients can be classified into two main categories: Type I and Type II. Type I occurs when the balance of endogenous heparin production and degradation is disturbed, with increased degradation of endogenous heparin by tumor-secreted heparanase. Type II hypercoagulability includes all the other etiologies, with factors related to the patient, the tumor, and/or the treatment. Patients with poor performance status are at higher risk of venous thromboembolism (VTE). Tumors can result in VTE through direct pressure on blood vessels, resulting in stasis. Several medications for cancer are correlated with a high risk of thrombosis. These include hormonal therapy (e.g., tamoxifen), chemotherapy (e.g., cisplatin, thalidomide and asparaginase), molecular targeted therapy (e.g., lenvatinib, osimertinib), and anti-angiogenesis monoclonal antibodies (e.g., bevacizumab and ramucirumab).
1. Introduction
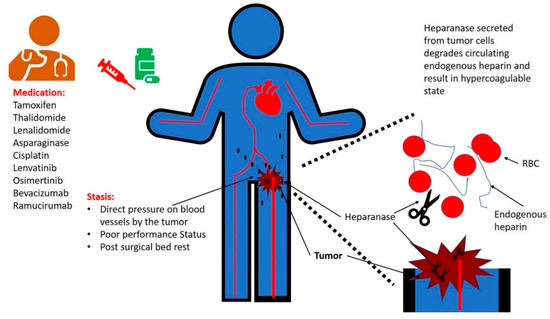
2. Stasis
3. Anti-Neoplastic Medications Associated with Increased Risk of Thrombosis
3.1. Tamoxifen
3.2. Chemotherapy
- (a)
-
Cisplatin. Cisplatin is associated with an increased risk of VTE and arterial thrombosis. A retrospective analysis from the Memorial Sloan Kettering Cancer Center found that 18.1% of cancer patients developed thrombosis during cisplatin treatment. Most of these cases (88%) occurred during the first 100 days from the initiation of cisplatin [19]. A meta-analysis of randomized controlled trials evaluating the incidence and risk of VTE associated with cisplatin-based chemotherapy showed a significantly increased risk of VTE with a relative risk of 1.67 [20]. VTE rates were 1.92% versus 0.79% in patients treated with cisplatin-based and non-cisplatin-based chemotherapy regimens, respectively [20]. A report from the UK National Cancer Research Institute of a randomized trial of patients with advanced gastroesophageal cancer randomized to epirubicin/(fluorouracil or capecitabine) and cisplatin or oxaliplatin found fewer thrombotic events in the oxaliplatin compared with the cisplatin groups, 7.6% vs. 15.1%, respectively; p = 0.0003 [21].
- (b)
-
Thalidomide. Thalidomide inhibits the production of interleukin (IL)-6, while suppressing proliferation and activating apoptosis of myeloma cells [22]. A study that treated patients with multiple myeloma using thalidomide and dexamethasone in preparation for autologous stem cell transplantation found VTE in 13% and 26% of patients treated with or without low-dose prophylactic warfarin, respectively [23]. A phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma showed that VTE occurred in 19.6% and 2.9% of patients treated with and without thalidomide, respectively [24].
- (c)
-
Asparaginase. Asparaginase is an enzyme that degrades L-asparagine, resulting in inhibition of protein synthesis in tumor cells [25]. A retrospective study reported thrombotic complications in adult patients with acute lymphoblastic leukemia receiving L-asparaginase during induction therapy in 4.2% of the patients [26]. A meta-analysis of 1752 patients from 17 prospective trials involving treatment with asparaginase demonstrated a rate of symptomatic thrombosis of 5.2% [27]. The UK ALL 2003 study reported asparaginase-related venous thrombosis in 3.2% of 1824 treated patients [28]. The use of prednisone and asparaginase concomitantly administered in a leukemic patient suffering from a prothrombotic risk factor (such as protein C deficiency, protein S deficiency, antithrombin deficiency, or factor V Leiden) was responsible for the onset of venous thrombosis in the majority of cases [29].
3.3. Molecular Targeted Therapies
- (d)
-
Lenvatinib is an oral medication that inhibits multiple receptor tyrosine kinases, including vascular endothelial growth factor receptors, fibroblast growth factor receptors, and platelet-derived growth factor receptor alpha [30]. A phase 2 trial treating patients with advanced, radioiodine-refractory thyroid cancer with lenvatinib, reported pulmonary embolism in 3% of the patients and DVT in 3% of the patients [31].
- (e)
-
Osimertinib is an epidermal growth factor receptor inhibitor that is implicated with an enhanced risk of thrombosis. The dose escalation study showed that pulmonary embolism occurred in 2.4% of the treated patients [32]. Osimertinib-induced VTE after initiation of osimertinib treatment was reported recently by Shiroyama et al. [33].
3.4. Anti-angiogenesis Monoclonal Antibodies
- (f)
-
Bevacizumab. Bevacizumab is a monoclonal antibody that targets vascular endothelial growth factor (VEGF) in the circulation. The addition of bevacizumab to irinotecan, fluorouracil, and leucovorin resulted in improvement in survival among patients with metastatic colorectal cancer; however, thrombotic events were higher in patients treated with bevacizumab compared to patients treated with chemotherapy alone (19.4% versus 16.2%, respectively, p = 0.26) [34]. Analysis of data pooled from five randomized controlled trials found that the combination of bevacizumab and chemotherapy, compared with chemotherapy alone, was associated with an increased risk of arterial thromboembolism with a hazard ratio of 2.0 [35]. A meta-analysis of 20 randomized controlled trials found that the incidence of arterial thrombotic events in patients receiving bevacizumab was 3.3% [36]. This meta-analysis showed the varying risk for arterial thrombotic events with different malignancies treated with bevacizumab, with the highest relative risk of 3.72 for patients with renal cell cancer, and with the relative risk being 1.89 in patients with colorectal cancer treated [36].
- (g)
-
Ramucirumab. Ramucirumab is a monoclonal antibody that targets the extracellular domain of VEGF receptor 2, and thus prevents its activation by VEGF [37]. A phase I pharmacologic and biologic study of ramucirumab reported DVT in 5.4% of the patients [37]. A study comparing ramucirumab versus placebo in combination with second-line chemotherapy in patients with metastatic colorectal carcinoma reported a nonsignificant difference in VTE of 8.2% and 6.3% with ramucirumab and placebo, respectively [38].
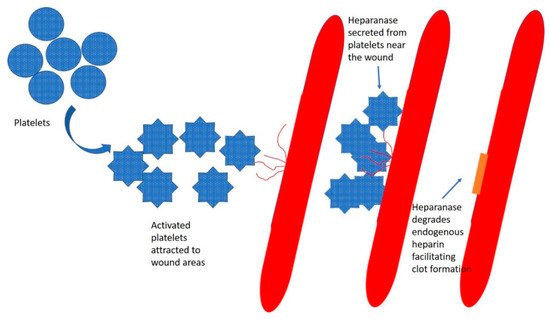
4. Heparin and Heparan Sulphate
5. Heparanase
References
- Trousseau, A. Phlegmasia alba dolens in: Lectures on Clinical Medicine, delivered at the Hôtel-Dieu, Paris; New Sydenham Society: London, UK, 1872; pp. 281–295.
- Dickson, B.C. Venous thrombosis: On the history of Virchow’s triad. Univ. Tor. Med. J. 2004, 81, 166–171.
- Nasser, N.J.; Sarig, G.; Brenner, B.; Nevo, E.; Goldshmidt, O.; Zcharia, E.; Li, J.P.; Vlodavsky, I. Heparanase neutralizes the anticoagulation properties of heparin and low-molecular-weight heparin. J. Thromb. Haemost. JTH 2006, 4, 560–565.
- Nasser, N.J.; Na’amad, M.; Weinberg, I.; Gabizon, A.A. Pharmacokinetics of low molecular weight heparin in patients with malignant tumors. Anti-Cancer Drugs 2015, 26, 106–111.
- Nadir, Y.; Brenner, B.; Zetser, A.; Ilan, N.; Shafat, I.; Zcharia, E.; Goldshmidt, O.; Vlodavsky, I. Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J. Thromb. Haemost. 2006, 4, 2443–2451.
- Nadir, Y.; Brenner, B.; Gingis-Velitski, S.; Levy-Adam, F.; Ilan, N.; Zcharia, E.; Nadir, E.; Vlodavsky, I. Heparanase induces tissue factor pathway inhibitor expression and extracellular accumulation in endothelial and tumor cells. Thromb. Haemost. 2008, 99, 133–141.
- Chiu, J.-J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387.
- Sigel, B.; Coelho, J.; Spigos, D.G.; Flanigan, D.P.; Schuler, J.J.; Kasprisin, D.O.; Nyhus, L.M.; Capek, V. Ultrasonography of blood during stasis and coagulation. Investig. Radiol. 1981, 16, 71–76.
- Fareed, J.; Walenga, J.M.; Kumar, A.; Rock, A. A modified stasis thrombosis model to study the antithrombotic actions of heparin and its fractions. Semin. Thromb. Hemost. 1985, 11, 155–175.
- White, C.; Noble, S.I.R.; Watson, M.; Swan, F.; Allgar, V.L.; Napier, E.; Nelson, A.; McAuley, J.; Doherty, J.; Lee, B.; et al. Prevalence, symptom burden, and natural history of deep vein thrombosis in people with advanced cancer in specialist palliative care units (HIDDen): A prospective longitudinal observational study. Lancet Haematol. 2019, 6, e79–e88.
- Metcalf, R.L.; Fry, D.J.; Swindell, R.; McGurk, A.; Clamp, A.R.; Jayson, G.C.; Hasan, J. Thrombosis in ovarian cancer: A case control study. Br. J. Cancer 2014, 110, 1118–1124.
- Beck-Razi, N.; Kuzmin, A.; Koren, D.; Sarig, G.; Brenner, B.; Haim, N.; Gaitini, D. Asymptomatic deep vein thrombosis in advanced cancer patients: The value of venous sonography. J. Clin. Ultrasound JCU 2010, 38, 232–237.
- Natsumeda, M.; Uzuka, T.; Watanabe, J.; Fukuda, M.; Akaiwa, Y.; Hanzawa, K.; Okada, M.; Oishi, M.; Fujii, Y. High Incidence of Deep Vein Thrombosis in the Perioperative Period of Neurosurgical Patients. World Neurosurg. 2018, 112, e103–e112.
- Chaichana, K.L.; Pendleton, C.; Jackson, C.; Martinez-Gutierrez, J.C.; Diaz-Stransky, A.; Aguayo, J.; Olivi, A.; Weingart, J.; Gallia, G.; Lim, M.; et al. Deep venous thrombosis and pulmonary embolisms in adult patients undergoing craniotomy for brain tumors. Neurol. Res. 2013, 35, 206–211.
- Osaki, T.; Saito, H.; Fukumoto, Y.; Kono, Y.; Murakami, Y.; Shishido, Y.; Kuroda, H.; Matsunaga, T.; Sato, K.; Hirooka, Y.; et al. Risk and incidence of perioperative deep vein thrombosis in patients undergoing gastric cancer surgery. Surg. Today 2018, 48, 525–533.
- Fisher, B.; Costantino, J.; Redmond, C.; Poisson, R.; Bowman, D.; Couture, J.; Dimitrov, N.V.; Wolmark, N.; Wickerham, D.L.; Fisher, E.R.; et al. A Randomized Clinical Trial Evaluating Tamoxifen in the Treatment of Patients with Node-Negative Breast Cancer Who Have Estrogen-Receptor–Positive Tumors. N. Engl. J. Med. 1989, 320, 479–484.
- Fisher, B.; Dignam, J.; Wolmark, N.; DeCillis, A.; Emir, B.; Wickerham, D.L.; Bryant, J.; Dimitrov, N.V.; Abramson, N.; Atkins, J.N.; et al. Tamoxifen and chemotherapy for lymph node-negative, estrogen receptor-positive breast cancer. J. Natl. Cancer Inst. 1997, 89, 1673–1682.
- Khorana, A.A.; Dalal, M.; Lin, J.; Connolly, G.C. Incidence and predictors of venous thromboembolism (VTE) among ambulatory high-risk cancer patients undergoing chemotherapy in the United States. Cancer 2013, 119, 648–655.
- Moore, R.A.; Adel, N.; Riedel, E.; Bhutani, M.; Feldman, D.R.; Tabbara, N.E.; Soff, G.; Parameswaran, R.; Hassoun, H. High incidence of thromboembolic events in patients treated with cisplatin-based chemotherapy: A large retrospective analysis. J. Clin. Oncol. 2011, 29, 3466–3473.
- Seng, S.; Liu, Z.; Chiu, S.K.; Proverbs-Singh, T.; Sonpavde, G.; Choueiri, T.K.; Tsao, C.-K.; Yu, M.; Hahn, N.M.; Oh, W.K. Risk of venous thromboembolism in patients with cancer treated with Cisplatin: A systematic review and meta-analysis. J. Clin. Oncol. 2012, 30, 4416–4426.
- Starling, N.; Rao, S.; Cunningham, D.; Iveson, T.; Nicolson, M.; Coxon, F.; Middleton, G.; Daniel, F.; Oates, J.; Norman, A.R. Thromboembolism in patients with advanced gastroesophageal cancer treated with anthracycline, platinum, and fluoropyrimidine combination chemotherapy: A report from the UK National Cancer Research Institute Upper Gastrointestinal Clinical Studies Group. J. Clin. Oncol. 2009, 27, 3786–3793.
- Anderson, K.C. Lenalidomide and thalidomide: Mechanisms of action-similarities and differences. Semin. Hematol. 2005, 42, S3–S8.
- Cavo, M.; Zamagni, E.; Tosi, P.; Cellini, C.; Cangini, D.; Tacchetti, P.; Testoni, N.; Tonelli, M.; de Vivo, A.; Palareti, G. First-line therapy with thalidomide and dexamethasone in preparation for autologous stem cell transplantation for multiple myeloma. Haematologica 2004, 89, 826–831.
- Rajkumar, S.V.; Blood, E.; Vesole, D.; Fonseca, R.; Greipp, P.R. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: A clinical trial coordinated by the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2006, 24, 431–436.
- Batool, T.; Makky, E.A.; Jalal, M.; Yusoff, M.M. A comprehensive review on L-asparaginase and its applications. Appl. Biochem. Biotechnol. 2016, 178, 900–923.
- Gugliotta, L.; Mazzucconi, M.G.; Leone, G.; Mattioli-Belmonte, M.; Defazio, D.; Annino, L.; Tura, S.; Mandelli, F.; Group, G. Incidence of thrombotic complications in adult patients with acute lymphoblastic leukaemia receiving L-asparaginase during induction therapy: A retrospective study. Eur. J. Haematol. 1992, 49, 63–66.
- Caruso, V.; Iacoviello, L.; Di Castelnuovo, A.; Storti, S.; Mariani, G.; De Gaetano, G.; Donati, M.B. Thrombotic complications in childhood acute lymphoblastic leukemia: A meta-analysis of 17 prospective studies comprising 1752 pediatric patients. Blood 2006, 108, 2216–2222.
- Qureshi, A.; Mitchell, C.; Richards, S.; Vora, A.; Goulden, N. Asparaginase-related venous thrombosis in UKALL 2003- re-exposure to asparaginase is feasible and safe. Br. J. Haematol. 2010, 149, 410–413.
- Nowak-Göttl, U.; Heinecke, A.; von Kries, R.; Nürnberger, W.; Münchow, N.; Junker, R. Thrombotic events revisited in children with acute lymphoblastic leukemia: Impact of concomitant Escherichia coli asparaginase/prednisone administration. Thromb. Res. 2001, 103, 165–172.
- Nishio, M.; Horai, T.; Horiike, A.; Nokihara, H.; Yamamoto, N.; Takahashi, T.; Murakami, H.; Yamamoto, N.; Koizumi, F.; Nishio, K.; et al. Phase 1 study of lenvatinib combined with carboplatin and paclitaxel in patients with non-small-cell lung cancer. Br. J. Cancer 2013, 109, 538–544.
- Cabanillas, M.E.; Schlumberger, M.; Jarzab, B.; Martins, R.G.; Pacini, F.; Robinson, B.; McCaffrey, J.C.; Shah, M.H.; Bodenner, D.L.; Topliss, D. A phase 2 trial of lenvatinib (E7080) in advanced, progressive, radioiodine-refractory, differentiated thyroid cancer: A clinical outcomes and biomarker assessment. Cancer 2015, 121, 2749–2756.
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L.; et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1689–1699.
- Shiroyama, T.; Hayama, M.; Satoh, S.; Nasu, S.; Tanaka, A.; Morita, S.; Morishita, N.; Suzuki, H.; Okamoto, N.; Hirashima, T. Successful retreatment with osimertinib after osimertinib-induced acute pulmonary embolism in a patient with lung adenocarcinoma: A case report. Respir. Med. Case Rep. 2016, 20, 25–27.
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342.
- Scappaticci, F.A.; Skillings, J.R.; Holden, S.N.; Gerber, H.-P.; Miller, K.; Kabbinavar, F.; Bergsland, E.; Ngai, J.; Holmgren, E.; Wang, J. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J. Natl. Cancer Inst. 2007, 99, 1232–1239.
- Ranpura, V.; Hapani, S.; Chuang, J.; Wu, S. Risk of cardiac ischemia and arterial thromboembolic events with the angiogenesis inhibitor bevacizumab in cancer patients: A meta-analysis of randomized controlled trials. Acta Oncol. 2010, 49, 287–297.
- Spratlin, J.L.; Mulder, K.E.; Mackey, J.R. Ramucirumab (IMC-1121B): A novel attack on angiogenesis. Future Oncol. 2010, 6, 1085–1094.
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.-E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015, 16, 499–508.
- Wardrop, D.; Keeling, D. The story of the discovery of heparin and warfarin. Br. J. Haematol. 2008, 141, 757–763.
- Howell, W. The purification of heparin and its presence in blood. Am. J. Physiol. Leg. Content 1925, 71, 553–562.
- Casu, B.; Lindahl, U. Structure and biological interactions of heparin and heparan sulfate. In Advances in Carbohydrate Chemistry and Biochemistry; Academic Press: Cambridge, MA, USA, 2001; Volume 57, pp. 159–206.
- Hacker, U.; Nybakken, K.; Perrimon, N. Heparan sulphate proteoglycans: The sweet side of development. Nat. Rev. Mol. Cell Biol. 2005, 6, 530–541.
- Weiss, R.J.; Esko, J.D.; Tor, Y. Targeting heparin and heparan sulfate protein interactions. Org. Biomol. Chem. 2017, 15, 5656–5668.
- Tremblay, J.F. Making heparin safe. Chem. Eng. News 2016, 94, 30–34.
- Kouta, A.; Jeske, W.; Hoppensteadt, D.; Iqbal, O.; Yao, Y.; Fareed, J. Comparative Pharmacological Profiles of Various Bovine, Ovine, and Porcine Heparins. Clin. Appl. Thromb. Hemost. Off. J. Int. Acad. 2019, 25.
- Kakkar, A.K.; Levine, M.N.; Kadziola, Z.; Lemoine, N.R.; Low, V.; Patel, H.K.; Rustin, G.; Thomas, M.; Quigley, M.; Williamson, R.C. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: The fragmin advanced malignancy outcome study (FAMOUS). J. Clin. Oncol. 2004, 22, 1944–1948.
- Lee, A.Y.; Levine, M.N.; Baker, R.I.; Bowden, C.; Kakkar, A.K.; Prins, M.; Rickles, F.R.; Julian, J.A.; Haley, S.; Kovacs, M.J.; et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N. Engl. J. Med. 2003, 349, 146–153.
- Lebeau, B.; Chastang, C.; Brechot, J.M.; Capron, F.; Dautzenberg, B.; Delaisements, C.; Mornet, M.; Brun, J.; Hurdebourcq, J.P.; Lemarie, E. Subcutaneous heparin treatment increases survival in small cell lung cancer. Cancer 1994, 74, 38–45.
- Lazo-Langner, A.; Goss, G.; Spaans, J.; Rodger, M. The effect of low-molecular-weight heparin on cancer survival. A systematic review and meta-analysis of randomized trials. J. Thromb. Haemost. 2007, 5, 729–737.
- Abboud-Jarrous, G.; Atzmon, R.; Peretz, T.; Palermo, C.; Gadea, B.B.; Joyce, J.A.; Vlodavsky, I. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J. Biol. Chem. 2008, 283, 18167–18176.
- Levy-Adam, F.; Miao, H.Q.; Heinrikson, R.L.; Vlodavsky, I.; Ilan, N. Heterodimer formation is essential for heparanase enzymatic activity. Biochem. Biophys. Res. Commun. 2003, 308, 885–891.
- Abboud-Jarrous, G.; Rangini-Guetta, Z.; Aingorn, H.; Atzmon, R.; Elgavish, S.; Peretz, T.; Vlodavsky, I. Site-directed mutagenesis, proteolytic cleavage, and activation of human proheparanase. J. Biol. Chem. 2005, 280, 13568–13575.
- Nasser, N.J. Heparanase involvement in physiology and disease. Cell. Mol. Life Sci. CMLS 2008, 65, 1706–1715.
- Nasser, N.J.; Avivi, A.; Shafat, I.; Edovitsky, E.; Zcharia, E.; Ilan, N.; Vlodavsky, I.; Nevo, E. Alternatively spliced Spalax heparanase inhibits extracellular matrix degradation, tumor growth, and metastasis. Proc. Natl. Acad. Sci. USA 2009, 106, 2253–2258.
- Freeman, C.; Parish, R.C. Human platelet heparanase: Purification, characterization and catalytic activity. Biochem. J. 1998, 330, 1341–1350.
- Eldor, A.; Bar-Ner, M.; Yahalom, J.; Fuks, Z.; Vlodavsky, I. Role of heparanase in platelet and tumor cell interactions with the subendothelial extracellular matrix. Semin. Thromb. Hemost. 1987, 13, 475–488.
- Tan, Y.X.; Cui, H.; Wan, L.M.; Gong, F.; Zhang, X.; Vlodavsky, I.; Li, J.P. Overexpression of heparanase in mice promoted megakaryopoiesis. Glycobiology 2018, 28, 269–275.
- Cui, H.; Tan, Y.X.; Osterholm, C.; Zhang, X.; Hedin, U.; Vlodavsky, I.; Li, J.P. Heparanase expression upregulates platelet adhesion activity and thrombogenicity. Oncotarget 2016, 7, 39486–39496.
- Goshen, R.; Hochberg, A.A.; Korner, G.; Levy, E.; Ishai-Michaeli, R.; Elkin, M.; de Groot, N.; Vlodavsky, I. Purification and characterization of placental heparanase and its expression by cultured cytotrophoblasts. Mol. Hum. Reprod. 1996, 2, 679–684.
- Vlodavsky, I.; Friedmann, Y.; Elkin, M.; Aingorn, H.; Atzmon, R.; Ishai-Michaeli, R.; Bitan, M.; Pappo, O.; Peretz, T.; Michal, I.; et al. Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nat. Med. 1999, 5, 793–802.
- Haimov-Kochman, R.; Friedmann, Y.; Prus, D.; Goldman-Wohl, D.S.; Greenfield, C.; Anteby, E.Y.; Aviv, A.; Vlodavsky, I.; Yagel, S. Localization of heparanase in normal and pathological human placenta. Mol. Hum. Reprod. 2002, 8, 566–573.
- Eddy, A.C.; Chapman, H.; George, E.M. Heparanase regulation of sFLT-1 release in trophoblasts in vitro. Placenta 2019, 85, 63–68.
- Hambruch, N.; Kumstel, S.; Haeger, J.D.; Pfarrer, C. Bovine placentomal heparanase and syndecan expression is related to placental maturation. Placenta 2017, 57, 42–51.
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162.
- Nasser, N.J.; Avivi, A.; Shushy, M.; Vlodavsky, I.; Nevo, E. Cloning, expression, and characterization of an alternatively spliced variant of human heparanase. Biochem. Biophys. Res. Commun. 2007, 354, 33–38.
- Nasser, N.J.; Nevo, E.; Shafat, I.; Ilan, N.; Vlodavsky, I.; Avivi, A. Adaptive evolution of heparanase in hypoxia-tolerant Spalax: Gene cloning and identification of a unique splice variant. Proc. Natl. Acad. Sci. USA 2005, 102, 15161–15166.
- Nasser, N.J.; Nevo, E. Heparanase patents: Dim past and bright future. Recent Pat. Inflamm. Allergy Drug Discov. 2013, 7, 162–167.
- Sandwall, E.; Bodevin, S.; Nasser, N.J.; Nevo, E.; Avivi, A.; Vlodavsky, I.; Li, J.P. Molecular structure of heparan sulfate from Spalax. Implications of heparanase and hypoxia. J. Biol. Chem. 2009, 284, 3814–3822.

