 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Barbara Jacczak | + 1499 word(s) | 1499 | 2021-06-23 06:03:06 | | | |
| 2 | Rita Xu | Meta information modification | 1499 | 2021-06-29 10:11:10 | | | | |
| 3 | Conner Chen | Meta information modification | 1499 | 2021-10-11 09:55:15 | | |
Video Upload Options
Proper functioning of cells—their ability to divide, differentiate, and regenerate—is dictated by genomic stability. The main factors contributing to this stability are the telomeric ends that cap chromosomes. All these processes are accompanied by telomere-length modulation. Maintaining the key levels of telomerase component (hTERT) expression and telomerase activity that provide optimal telomere length as well as some nontelomeric functions represents a promising step in advanced anti-aging strategies, especially in dermocosmetics. Therefore telomere biology and telomerase activity have been of interest to scientists in various medical science fields for years, including the study of both cancer and of senescence and aging. This paper focuses on the metabolic potential of natural compounds to modulate telomerase and telomere biology and thus prevent senescence and skin aging.
1. Introduction
2. Molecular Basis of Senescence and Aging
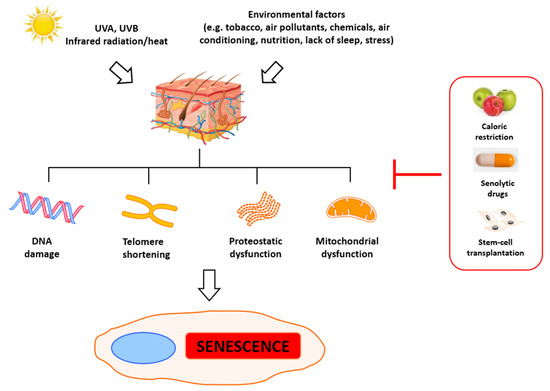
3. Roles of Telomeres in Skin Aging
The primary indicators of aging are telomere attrition, epigenetic and mitochondrial dysfunction, and general DNA damage. Significantly, this process can be accelerated by stress, which leads to the accumulation of damage and eventually to mitotic crisis orcell death [26]. Numerous agents can trigger DNA damage, including ionizing radiation, radiomimetic drugs, reactive oxygen species, metabolic errors during replication and transcription [27], and deficient DNA repair [28]. Some of those factors are just a result of cells’ metabolic activity and cannot be eliminated, but some of them can be prevented to slow down senescence and aging. Evaluation of the aging of skin has revealed the primary roles of alterations observed in the metabolism of fibroblasts. In young skin, fibroblasts adhere to the surrounding intact extracellular matrix (ECM), which is mainly comprised of type I collagen [29][30]. In aged skin, fibroblast attachment is impaired due to progressive ECM degradation, resulting in fibroblast size reduction, decreased elongation, and collapsed morphology [29][31][32][33][34]. Simultaneously, their proliferative potential is also significantly reduced, which impedes skin regeneration, thus contributing to progressive aging. The reduction of dermal fibroblast spreading and cell size can also increase mitochondrial ROS generation, which induces senescence [35].
References
- Quan, C.; Cho, M.K.; Perry, D.; Quan, T. Age-associated reduction of cell spreading induces mitochondrial DNA common deletion by oxidative stress in human skin dermal fibroblasts: Implication for human skin connective tissue aging. J. Biomed. Sci. 2015, 22, 62.
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247.
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309.
- Attia, E.A.S.; Seada, L.S.; El-Sayed, M.H.; El-Shiemy, S.M. Study of telomerase reverse transcriptase (hTERT) expression in normal, aged, and photo-aged skin. Int. J. Dermatol. 2010, 49, 886–893.
- Liu, J.-P.; Wang, L.; Wang, Z. Roles of Telomere Biology in Cell Senescence, Replicative and Chronological Ageing. Cells 2019, 8, 54.
- Haendeler, J.; Dröse, S.; Büchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial Telomerase Reverse Transcriptase Binds to and Protects Mitochondrial DNA and Function From Damage. Arter. Thromb. Vasc. Biol. 2009, 29, 929–935.
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130.
- Ferrucci, L.; Gonzalez-Freire, M.; Fabbri, E.; Simonsick, E.; Tanaka, T.; Moore, Z.; Salimi, S.; Sierra, F.; De Cabo, R. Measuring biological aging in humans: A quest. Aging Cell 2020, 19, e13080.
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435.
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77.
- Campisi, J.; Daddadifagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740.
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479.
- Lo’pez-Otı’, C. The Hallmarks of Aging. Cell 2013, 153, 2013.
- Hodes, R. Disease drivers of aging. Ann. N. Y. Acad. Sci. 2016, 1386, 45–68.
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621.
- Damiani, E.; Brugè, F.; Cirilli, I.; Marcheggiani, F.; Olivieri, F.; Armeni, T.; Cianfruglia, L.; Giuliani, A.; Orlando, P.; Tiano, L. Modulation of Oxidative Status by Normoxia and Hypoxia on Cultures of Human Dermal Fibroblasts: How Does It Affect Cell Aging? Hindawi Oxidative Med. Cell. Longev. 2018, 2018, 5469159.
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011.
- Chandrasekaran, A.; Idelchik, M.D.P.S.; Melendez, J.A. Redox control of senescence and age-related disease. Redox Biol. 2017, 11, 91–102.
- Wang, A.S.; Dreesen, O. Biomarkers of Cellular Senescence and Skin Aging. Front. Genet. 2018, 9, 247.
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene. 2013, 32, 5129–5143.
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic. Acids Res. 2014, 42, 7666–7680.
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420.
- Ohtani, N.; Yamakoshi, K.; Takahashi, A.; Hara, E. The p16INK4a-RB pathway: Molecular link between cellular senescence and tumor suppression. J. Med. Investig. 2004, 51, 146–153.
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797.
- Fumagalli, M.; Rossiello, F.; Mondello, C.; d’Adda di Fagagna, F. Stable Cellular Senescence Is Associated with Persistent DDR Activation. PLoS ONE 2014, 9, e110969.
- Hönigsmann, H.; Schuler, G.; Aberer, W.; Romani, N.; Wolff, K. Immediate Pigment Darkening Phenomenon. A Reevaluation of Its Mechanisms. J. Investig. Dermatol. 1986, 87, 648–652.
- Kang, S.M.; Han, S.; Oh, J.-H.; Lee, Y.M.; Park, C.-H.; Shin, C.-Y.; Lee, D.H.; Chung, J.H. A synthetic peptide blocking TRPV1 activation inhibits UV-induced skin responses. J. Dermatol. Sci. 2017, 88, 126–133.
- Aubert, G.; Lansdorp, P.M. Telomeres and Aging. Physiol. Rev. 2008, 88, 557–579.
- Pani, B.; Nudler, E. Mechanistic Insights into Transcription Coupled DNA Repair. Author manuscript; available in PMC 2018 August 01. DNA Repair 2017, 56, 42–50. [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nat. Cell Biol. 2014, 509, 439–446.
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol.2016, 231, 3–14. [CrossRef] [PubMed]
- Fisher, G.J.; Varani, J.; Voorhees, J.J. Looking older: Fibroblast collapse and therapeutic implications. Arch. Dermatol. 2008, 144, 666–672. [CrossRef] [PubMed]
- Cole, M.A.; Quan, T.; Voorhees, J.J.; Fisher, G.J. Extracellular matrix regulation of fibroblast function: Redefining our perspective on skin aging. J. Cell Commun. Signal. 2018, 12, 35–43. [CrossRef] Int. J. Mol. Sci. 2021, 22, 6381 18 of 22
- Varani, J.; Dame, M.K.; Rittie, L.; Fligiel, S.E.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Decreased Collagen Production in Chronologically Aged Skin: Roles of Age-Dependent Alteration in Fibroblast Function and Defective Mechanical Stimulation. Am. J. Pathol. 2006,168, 1861–1868.
- El-Domyati, M.; Attia, S.; Saleh, F.; Brown, D.; Birk, D.E.; Gasparro, F.; Ahmad, H.; Uitto, J. Intrinsic aging vs. photoaging: A comparative histopathological, immunohistochemical, and ultrastructural study of skin. Exp. Dermatol. 2002, 11, 398–405.

