Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nicolai Ostberg | + 4380 word(s) | 4380 | 2021-06-03 10:57:19 | | | |
| 2 | Vivi Li | Meta information modification | 4380 | 2021-06-28 03:59:20 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ostberg, N.; Zafar, M. Thoracic Aortic Aneurysms and Dissection. Encyclopedia. Available online: https://encyclopedia.pub/entry/11331 (accessed on 05 June 2026).
Ostberg N, Zafar M. Thoracic Aortic Aneurysms and Dissection. Encyclopedia. Available at: https://encyclopedia.pub/entry/11331. Accessed June 05, 2026.
Ostberg, Nicolai, Mohammad Zafar. "Thoracic Aortic Aneurysms and Dissection" Encyclopedia, https://encyclopedia.pub/entry/11331 (accessed June 05, 2026).
Ostberg, N., & Zafar, M. (2021, June 26). Thoracic Aortic Aneurysms and Dissection. In Encyclopedia. https://encyclopedia.pub/entry/11331
Ostberg, Nicolai and Mohammad Zafar. "Thoracic Aortic Aneurysms and Dissection." Encyclopedia. Web. 26 June, 2021.
Copy Citation
Thoracic aortic aneurysm and dissection (TAAD) affects many patients globally and has high mortality rates if undetected. Once thought to be solely a degenerative disease that afflicted the aorta due to high pressure and biomechanical stress, extensive investigation of the heritability and natural history of TAAD has shown a clear genetic basis for the disease.
thoracic aortic aneurysm and dissection (TAAD)
genetics
genetic screening
syndromic TAAD
non-syndromic TAAD
1. Introduction
Thoracic aortic aneurysm (TAA) is a dangerous, deadly, and silent disease that is notoriously difficult to detect and diagnose prior to complications [1]. It is believed that only 5% of TAA are symptomatic prior to dissection or rupture and even when dissection presents with symptoms, less than 50% are promptly diagnosed in the emergency department prior to death [2]. Together, TAA and abdominal aortic aneurysms (AAA) constitute the 17th leading cause of death in patients over the age of 65 [3]. Furthermore, many TAA dissections tend to be misdiagnosed as myocardial infarctions, suggesting a higher true mortality rate [4]. Yet, if TAAs are diagnosed and managed surgically prior to dissection, survival nearly matches population controls, with limited complications [5][6]. Therefore, early diagnosis and management remain a critical component for limiting mortality from TAA.
Due to the difficulty of diagnosing TAA based on symptoms alone, efforts have been undertaken to determine which patients are at higher risk of harboring TAA and therefore, warrant preemptive screening for early diagnosis of aneurysm, monitoring, and appropriately tuned surgical intervention [7]. Because more than 20% of patients report a positive family history, the genetics of thoracic aortic aneurysm and dissection (TAAD) has been extensively investigated as a potential avenue for both diagnosis and risk stratification [8].
Traditionally, TAAD is divided into syndromic—with other organ system abnormalities other than the aorta—and non-syndromic—with no other systemic abnormalities present. Non-syndromic disease is then further subdivided to include familial non-syndromic TAAD, where one or more family members are diagnosed with TAAD, and non-familial TAAD, where no other family members are affected [9]. However, in both syndromic and non-syndromic cases, often, only a single gene is mutated and that same mutated gene can produce both syndromic and non-syndromic TAAD [10]. Moreover, there is significant variation in the severity of TAAD presentation, even within the same family [11]. Therefore, this classification system provides a convenient hierarchy for discussing the genetic causes of TAAD, although the true clinical picture is much more complex.
Several causes of syndromic TAAD are well described, such as Marfan syndrome (MFS), Loeys–Dietz syndrome (LDS), and Ehlers-Danlos Syndrome (EDS) [12]. While the existence of systemic features may make diagnosis easier, the wide spectrum of physical manifestations that these diseases can assume makes the task of prompt diagnosis more difficult. A high index of suspicion must be maintained to detect the more mild forms of syndromic manifestations or presentations that overlap with other conditions [13][14]. The prognosis of syndromic TAA cases is generally worse than non-syndromic cases, resulting in modified surgical intervention criteria at aortic diameters less than the typical boundary of a 5.0–5.5 cm aortic diameter [15]. Regardless, syndromic TAAD accounts for only approximately one-fifth of all TAAD cases, meaning that other genes and mechanisms must be implicated in the development of TAAD [2].
Cases of non-syndromic TAA are more prevalent, but identifying these patients can be challenging. Some cases can be explained by mutations in genes involved in syndromic TAAD [16]. Approximately 20% of non-syndromic patients have one affected family member, known as familial TAAD, providing further evidence for a genetic link [17][18][19]. However, this leaves a substantial majority of patients with no cause of syndromic TAAD and no family history—what can be thought of as “random” or sporadic TAAD. More investigation is being undertaken to understand these seemingly random cases of TAAD and some interesting associations have been uncovered. For example, mutations in the causative gene in MFS have also been linked to sporadic TAAD [20]. However, further work needs to be done to fully understand the genetic interactions that lead to both familial and sporadic TAAD.
2. Causes of Syndromic TAAD
Syndromic conditions that cause an increased risk of TAAD have been well described for decades, although the causative mutations have only just recently been verified. Typically, syndromic TAAD is linked to dysfunction of the extracellular matrix (ECM) [21], medial smooth muscle cells (SMC) [22], or TGF- β signaling [23]. While several other pathologies are often simultaneously present in these syndromic cases, the most deadly concern typically is vascular, as the weakening of the wall of the aorta can cause dilation, dissection, and potential rupture of the aorta. The three most common and well described diseases linked to syndromic TAAD and their respective pathophysiology are reviewed. Other syndromic causes are briefly discussed.
2.1. Marfan Syndrome (MFS)
Microfibrils, which are largely composed of two large glycoproteins, fibrilin-1 and fibrilin-2, constitute a structurally significant component for the integrity of the extracellular matrix throughout the body [24]. In association with essential elastin, microfibrils contribute significantly to the stability and elasticity of the aorta [25]. Mutations in the gene for fibrillin-1, FBN1, have been causatively linked to MFS, an autosomal dominant connective tissue disorder with numerous associated pathologies, including skeletal, ocular, and, of note for this review, cardiovascular abnormalities [26]. Missense mutations in FBN1 were first linked to MFS in 1991 [27]. Knowledge has since grown to include 1847 highly penetrant unique mutations, and it is likely that many more exist (http://www.umd.be/FBN1/—last updated 28 August 2014). Most mutations in FBN1 are heterozygous but recent evidence suggests that both homozygous and compound heterozygous mutations exists in approximately 0.5% of MFS patients [28]. It is important to note that FBN1 mutations have been shown to cause both syndromic (MFS) and non-syndromic TAAD; interestingly, variants in FBN1 were the most frequently mutated gene in a large whole exome sequencing study of syndromic and non-syndromic TAAD patients (Figure 1) [29].
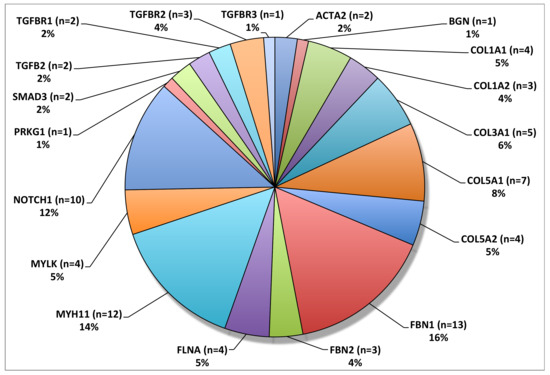
Figure 1. Overall frequency distribution of TAAD associated variants. FBN1 was the most frequently mutated gene in this patient cohort. © 2020 by Edi.Ermes. [30]
Investigation into these numerous mutations has revealed some genotype–phenotype associations. However, there is still a wide range of phenotypic variability in patients with the same FBN1 mutation, suggesting that complex gene interactions are at play [31]. Distinctions have been drawn between haploinsufficient (HI) mutations and dominant negative (DN) mutations. HI mutations of FBN1 result in only one functional copy of the gene, often the result of frameshift, nonsense, or splice-site mutation. The fibrillin levels decrease, resulting in reduced structural integrity of microfibrils [32]. Dominant negative (DN) mutations that result in a mutated protein lead to large phenotypic variability, depending on the region of the protein that is affected [33]. Evidence suggests that neither mutation class fully captures all of the phenotypic variation seen in FBN1 mutations; both HI and DN mutations cause MFS. However, patients with HI mutations have been shown to have significantly higher rates of aortic dissection, although overall cardiovascular mortality is comparable between the two mutation classes [34]. In addition, DN mutations in functionally significant cystine residues have been linked to higher probability of aortic dilation and dissection [35][36] and those affecting calcium binding exert structural effects on fibrillin assembly [37]. Interestingly, mutations in FBN1 have also been found in about 4% of TAAD patients without typical clinical manifestations of MFS [38]. Recent work has shown that FBN1 hypomorphic mice also displayed similar phenotypes, with increased elastin fragmentation, reduced wall strain, and larger aortic diameters [39].
Fibrilin-1 has also been shown to interact with TGF-β (transforming growth factor-β). TGF-β is a peptide that associates with latency-associated protein (LAP) to form the small latent complex, which, in turn, associated with latency-associated TGF-β binding protein to form the large latent complex (LLC). The LLC targets TGF-β to fibrillin microfibrils, sequestering it within the walls of the aorta [40]. It has been suggested that FBN1 mutations seen in MFS result in impaired TGF-β sequestering [41]. Because increased TGF-β signalling has been linked to TAAD [42], fibrillin-1′s inability to adequately regulate TGF-β may also be a causative or contributing factor to TAAD in MFS patients.
Clinically, patients presenting with MFS are at high risk for developing aortic dissections due to the structural instability of their aortas. MFS patient with FBN1 mutations are also more likely to develop dissections at the aortic root [43]. As with patients without MFS, the rate of dissection tends to correlate with diameter. The annual risk of dissection is 13% in MFS patients with aortic diameters greater than 5.0 cm [44]. The growth rate of these aneurysms has been measured at 0.26 cm/year, and rate of growth increases at larger diameters [44]. Currently, guidelines suggest operation when the aneurysm reaches 4.5–5.0 cm in diameter in MFS patients. Currently, clinical management of MFS patients does not typically vary based on the specific mutation present, due to the aforementioned high degree of phenotypic and behavioral variability across the same genetic mutation. However, certain groups of patients may benefit from surgery at smaller diameters: patients with rapidly growing aneurysms or patients who plan on becoming pregnant are suggested to receive prophylactic aortic repair surgery [45].
In the future, it is likely that surgical treatment for MFS patients will be predicated on the specific FBN1 mutation present, achieving an even higher level of genetically based precision medicine.
2.2. Loeys–Dietz Syndrome (LDS)
First described in 2005, Loeys–Dietz syndrome (LDS) is an autosomal dominant disorder characterized by severly increased risk of aortic aneurysms and dissection along with other defects [46]. Although the disease was first thought to be linked to mutations in the transforming growth factor β receptor I (TGFBR1) and transforming growth factor β receptor II (TGFBR2), other mutations have also been linked to LDS, including decapentaplegic homolog 3 (SMAD3) and transforming growth factor β II (TGFB2) [46][47][48]. Due to phenotypic variation across these different mutations and the high risk of aneurysm even without other associated systemic features of LDS, four different subgroups of LDS (LDS1-LDS4) were developed that describe different pathophysiologies and prognoses [49]. LDS1 and LDS2 are the two most common subtypes of LDS and encompass mutations in TGFBR1 and TGFBR2, respectively. Less studied, yet still important, are mutations in SMAD and TGFB2, which lead to LDS3 and LDS4, respectively [49].
TGF-β signaling plays a critical role in blood vessel development, cell differentiation, proliferation, homeostasis, and vascular maintenance [50] and all four subtypes of LDS have been shown to affect TGF-β signaling. Significant volumes of research have investigated the molecular mechanisms regarding how alterations in TGF-β pathways lead to aneurysms [51]. Paradoxically, although many of the TGFBR1/2 mutations seen in LDS1/2 are predicted to reduce TGF-β activity, the opposite is seen. High levels of byproducts of TGF-β signaling, such as increased SMAD2 phosphorylation, have been observed in patients with LDS1/2 [52]. Some hypotheses have been suggested to explain this effect, including that a dysregulated negative feedback loop through a non-canonical TGF-β pathway or excess signaling through an intact TGFBR homodimer. However, more research is needed to definitively explain these fluctuations in TGF-β signaling as they relate to aneurysm development. See the following reviews for extensive investigation of this paradox [42][53][54].
Mutations in SMAD3, the intracellular effectors of the TGF-β pathway, have been causatively linked to LDS3. 67 pathogenic mutations of SMAD3 have been identified. In total, 61% of SMAD3 mutations linked to LDS are missense mutations, followed by frameshift mutations (23%) [54]. Upon phosphorylation by TGFBR1, SMAD3 transmits signals to the nucleus upon association with SMAD4. Similar to other forms of LDS, mutations in SMAD3 have been associated with increased TGF-β signaling, as demonstrated by increased levels of nuclear translocation of SMAD3 and connective tissue growth factor (CTGF) [47].
The clinical course of LDS patients tends to be more severe than patients with MFS. Patients with LDS1/2 have had significant vascular complications reported at a young age, some within the first year of life, significant risk of dissection below 5.0 cm aortic diameter, and high mortality with a median life expectancy of 37 years [46]. Guidelines for the management of LDS have thus recommended surgical intervention between 4.0 and 4.2 cm, although recent commentary has suggested that this may be too aggressive [15].
2.3. Ehlers-Danlos Syndrome (EDS)
Mutations in the collagen type III, alpha 1 gene (COL3A1), when associated with syndromic features such as facial dysmorphia, visible veins through translucent skin, and easy bruising, are common manifestations of EDS type IV, otherwise known as vascular EDS (vEDS) [55]. vEDS has been shown to be inherited in an autosomal dominant manner. It is 100% penetrant, and accounts for 5–10% of all EDS cases [56][57]. Over 700 unique mutations of COL3A1 associated with vEDS have been identified, 50% of which are believed to be de novo mutations [58][59]. vEDS is rare, with an estimated prevalence of 1 in 150,000, but the high frequency of vascular complications means that morbidity and mortality are high [60]. Around 0.5–1% of patients develop unruptured aneurysms and an additional 4% develop intercranial hemorrhages [61][62]. Aortic dissection accounts for approximately 22% of all deaths in patients with vEDS, and these dissections were fatal 68% of the time [63].
The molecular mechanism of EDS is linked to its causative genetic mutation: alterations in the amount of functional collagen that is present. The main functional elements within type III collagen are [Gly-X-Y]343 repeats within triple helical regions; missense glycine substitutions in this region result in non-functional, dominant negative mutations [64]. Other mutations, such as splice-site variants, frame-shift, nonsense, and deletions have also been linked to EDS [63][65]. Mouse models of COL3A1 mutant mice revealed nearly absent collagen in media of the aorta and approximately one-third the number collagen fibrils in the adventitia compared to wild type [66]. As a result of these mutations, collagen assembly in arteries and the GI system fails, limiting accommodation to mechanical stress, which increases rupture and dissection risk [67]. Gastrointestinal and other organ rupture is also comorbid due to the same pathophysiology [68].
The clinical course of patients with EDS is fraught with complications from unstable vasculature prone to rupture. The median survival for all patients with EDS is 51, with females living slightly longer than males [63]. The median age of first complication is 29 years old and 80% of patients with EDS will experience a complication (vascular or GI) prior to the age of 40 [57][69]. Genetics have shed some light on patients who tend to have better outcomes from EDS. Contrarily to previous belief, individuals with HI mutations may actually have more prevalent aortic complications compared to glycine substitutions or other splice site variants [59][65][70]. Patients with mutations in the N or C terminus of COL3A1 or missense variants in the triple helix region may have milder cases of EDS but their vascular risk may remain high [69]. However, substantial heterogeneity between patients with identical mutations hampers clear genotype–phenotype associations that can be used in clinical practice [56]. Prognostication is further complicated by the fact that dissection can occur in patients with EDS can occur at midsized arterial diameters without aneurysm development, prompting a recommendation for prophylactic surgery at aortic diameters between 4.5 cm and 5.0 cm [71].
2.4. Other Causes of Syndromic TAAD
Several other genes have been linked to developing cases of syndromic TAAD, including BGN (Meester-Loeys syndrome) [72], EFEMP2 (Cutis laxa, AR type Ib) [73][74], ELN (Cutis laxa, AD type) [75], and SLC2A10 (Arterial tortuosity syndrome) [76] among others. While the molecular and cellular pathophysiologies of these conditions are complex and diverse, patients with these conditions do not consistently experience dissection or rupture at aortic diameters less than the recommended 5.0 cm aortic diameter for surgical intervention [71]. Therefore, from a clinical standpoint, these patients are not as concerning as the aforementioned syndromic causes and do not require accelerated, aggressive surgical management.
3. Causes of Non-Syndromic TAAD
Understanding the causes of non-syndromic TAAD has advanced significantly after decades of study. Because 21% of TAAD patients have another family member that is affected, positive family history significantly increases the severity of TAAD and the likelihood of aortic dissection (Figure 2). A genetic link for such cases was assumed and extensively investigated [18][77]. Importantly, this 21% estimate of heritability is likely an underestimate due to a lack of routine imaging that detects non-symptomatic aneurysms in family members [78]. Those without family members afflicted by TAAD—termed sporadic TAAD—were once thought to be entirely degenerative and not genetically mediated. However, the current research suggests that there may also be genetic mechanisms underlying sporadic TAAD due to variably penetrant gene expression [79]. Moreover, for many of these seemingly isolated cases, the proband may be the first in a subsequent line of inheritance. Other potential reasons why family history of TAAD might go unnoticed include asymptomatic aneurysms not detected or diagnosed, small families with few family members with which to trace inheritance over time, and a lack of sharing of health information amongst family members.
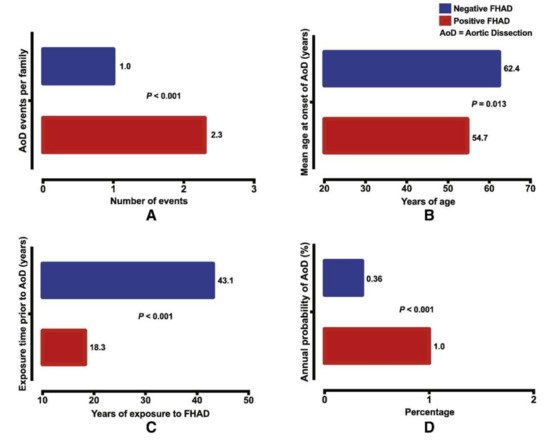
Figure 2. Positive family history of aortic dissection (FHAD) significantly increases the risk of developing a new dissection in unaffected family members, with (A) higher number of dissection events; (B) a younger age at dissection; (C) shorter duration of exposure prior to dissection; and (D) higher annual probability of aortic dissection. © 2020 by Elsevier. [77]
Familial TAAD appears to be more virulent than sporadic TAAD: age at first presentation is younger (58.2 vs. 65.7) and growth rates are faster (0.21 vs. 0.16 cm/year) [8] (Figure 2). Sporadic TAAD accounts for approximately 80% of all TAAD cases [80]. The ten-year mortality rate for non-syndromic TAAD is comparable to MFS (8.7% vs. 7.8%, respectively) [81]. Notably, genetic causality between syndromic and non-syndromic disease is difficult to separate; genes linked to syndromic TAAD (for example, FBN1 as previously discussed) are also known to cause non-syndromic TAAD [82] without other systemic manifestations of the mutation. Several genes have been linked to familial non-syndromic TAAD. Four play a key role in contraction of the vascular smooth muscles: ACTA2, MYLK, MYH11, and PRKG1 [83][84]. Similarly to the causes of syndromic TAAD, the mutations in these four genes have been demonstrated to produce dissections at aortic diameters <5.0 cm [85].
Sporadic TAAD remains a challenge to detect, diagnose, and manage, as many cases do not have as clear a link to genetic causes as familial or syndromic TAAD. Approximately 10% of early age sporadic aortic dissection cases have a mutation in causative syndromic or familial TAAD genes [79]. In total, 28% of cases had one or more variants of unknown significance [79]. Lifestyle factors such a hypertension also seem to play a significant role in sporadic TAA [86].
Here, we review the aforementioned four genes that cause TAAD at aortic diameters <5.0 cm and briefly discuss other genes that have been linked to non-syndromic TAAD and sporadic TAAD.
3.1. ACTA2
ACTA2 encodes SMC specific α-actin, which polymerizes to form thin filaments in the smooth muscle contraction element [87]. ACTA2 is the most common mutation linked to familial TAAD, with about 50% penetrance and accounts for between 12% and 21% of all cases seen, by far the most common genetic defect linked to familial TAAD to date [83][84][88][89]. The cumulative risk of aortic dissection or aortic repair by 85 years old is 76%, approaching rates seen in cases of syndromic TAAD [90]. The median age of dissection is 35 years old, and more type A dissections are found compared to type B (54% vs. 24%) [90]. However, unlike other familial TAAD pathologies, penetrance does not seem to vary with age [88].
A majority of the ACTA2 mutations that have been identified are DN mutations that prevent proper polymerization of actin or ATP hydrolysis that mediates SMC contraction [83]. Missense and premature truncating mutations have also been identified [83][91]. Aortic tissues in ACTA2 mutant patients are characterized by proteoglycan accumulation, decreased and disorganized SMCs, and fragmented elastic fibers [83]. Increased insulin growth factor 1 (IGF-1) expression has also been noted, possibly as a compensatory mechanism from the instability that ACTA2 mutations cause [92]. Patients also tend to have higher rates of occlusive vessel disease, leading to strokes and coronary artery disease, and one mutation has been linked to multisystemic smooth muscle dysfunction [89][93]. This evidence suggests a widespread and systemic disruption in adaptation to vascular wall stress, leading to aneurysm and dissection among other pathologies [94]. Surprisingly, mouse models have shown that simply deleting ACTA2 does not lead to aneurysm [95]; aneurysms developed only upon exposure to angiotensin II, indicating an interplay between genetics and environmental factors in the development of TAAD [96].
Notably, ACTA2 mutations tend to cause aortic dissection without preexisting aneurysm. As a result, one third of patients with ACTA2 mutations have acute Type A dissections at aortic diameters <5.0 cm [97]. Pregnant women with ACTA2 mutants are at even higher risk of small-diameter dissection, with dissections occurring between 3.8 and 4.7 cm in one study [97]. Patients with ACTA2 mutations are therefore recommended for surgical intervention between 4.5 and 5.0 cm.
3.2. MYLK
Myosin light chain kinase (MLCK), encoded by MYLK, is another gene that has been causatively linked to non-syndromic TAAD. MLCK is known to phosphorylate regulatory light chain (RLC), which, in turn, increases myosin II ATPase activity, endocytosis, and stress fiber response [98]. Phosphorylation of RLC by MLCK also influences SMC contraction and allows for fine control of arterial pressure [99].
Two MYLK mutations were shown to produce a significant reduction in MLCK activity via a HI mechanism that decreased MLCK activity by 85%, resulting in impaired SMC contraction [98]. Further mechanistic studies in mice linked a 50% reduction of MLCK activity to a 40% reduction in RLC phosphorylation and associated contraction of the aorta [100]. Although most vessels are able to withstand a 40% reduction in contraction capacity, the aorta is exposed to the highest forces in the body, making it the first vessel to demonstrate a pathological phenotype [100]. In terms of genotype-phenotype correlations, missense mutations were linked to a higher risk of Type A dissection compared to null mutations [101]. Another family with a MYLK mutation showed differential phenotypes between heterozygous and homozygous patients [102]. MYLK mutations associated with TAAD have lower penetrance compared to other syndromic and non-syndromic mutations; only 38% of mutant individuals in one study experienced aortic events [101].
Due to a high risk of dissection at diameters <5.0 cm, the current guidelines suggest surgical intervention between 4.5 and 5.0 cm. Further prospective validation regarding this suggestion is needed.
3.3. MYH11
MYH11 encodes smooth muscle myosin heavy chain (SM-MHC), a contractile component produced by SMCs [103]. Much like mutations in ACTA2, mutations in MYH11 results in dysregulation of SMC contraction, which is critical for maintaining aortic wall stability.
MYH11 mutations are thought to cause 1% of all familial TAAD cases [104]. This is inherited in an autosomal dominant fashion [9][105]. Specific mutations in the C-terminal coiled-coil region of SM-MHC have been linked to TAAD [103] and evidence suggests a DN mechanism [105]. The phenotype of these mutations—increased pulse wave velocity and decreased aortic compliance—demonstrates decreased elasticity in the aorta, which is linked to SMC dysfunction, and eventually leads to dissection [103]. Histological analysis of patients with MYH11 mutations reveals focal fibromuscular dysplasia in the vaso vasorum, which may be suggestive of an inflammatory immune response consistent with what has been observed in other TAAs [104][106]. In addition, dysregulation of IGF-1 and angiotensin II signaling suggests a distinct mechanism from other TGF-β related mutations and may explain the increased incidence of occlusive vascular pathology observed in these patients; specifically, IGF-1 signaling has been linked to both increased production of contractile proteins in SMCs and SMC proliferation [104]. One study also demonstrated that MYH11 mutations did not segregate with TAAD in one Dutch pedigree, suggesting that other unidentified genetic modifiers may play a role in the development of aneurysm [105]. Currently, surgical intervention is suggested at 4.5–5.0 cm [85][104].
3.4. PRKG1
PRKG1 encodes the type I cGMP-dependent protein kinase (PKG-1), which controls SMC contraction by regulating phosphatases that dephosphorylate the RLC [87]. The major isoform of PKG-1 that is present in SMCs, PKG-1α, is activated by increased cGMP levels [107]. A gain-of-function mutation associated with the cGMP binding site (Arg177Gln) structurally changes PKG-1α to the point where its inhibitory domain is no longer active, leading to continuous activation [108]. This activation results in decreased RLC phosphorylation, relaxing SMCs [87]. Recently, a case report revealed another PRKG1 mutation associated with the ATP binding domain that was also associated with aortic dissection [109].
Clinical data on PRKG1 mutations are limited but suggest a devastating aortic phenotype. In the one variant that has been identified to cause TAAD, penetrance of dissection is high (63%) and aortic enlargement was seen as good (37%). The average age at dissection was 37 years old, with the youngest occurring at 17 [108]. Similar to ACTA2, widespread vascular disease was noted in patients, including abdominal aneurysms, coronary aneurysms, coronary dissections, and tortuosity of the thoracic aorta [108]. Of the two patients with aortic diameters measured at the time dissection, one was noted at 4.3 cm [108]. Therefore, limited data notwithstanding, patients with PRKG1 are encouraged to have prophylactic surgery at 4.5–5.0 cm.
3.5. Other Genes Associated with Familial TAAD
Many other genes have been implicated in familial TAAD. These include the forkhead transcription factor (FOXE3) [110], lysyl oxidase (LOX) [111], methionine adenosyltransferase II α (MAT2A) [112], microfibril-associated glycoprotein 2 (MFAP5) [113], and notch homolog 1 (NOTCH1) [114]. Although patients with mutations in these genes tend to be at significantly higher risk for aortic aneurysm, they tend to follow the typical clinical course of aortic dilation followed by dissection at aortic diameters >5.0 cm. Therefore, standard guidelines regarding surgical intervention at aortic diameters between 5.0 and 5.5 cm are recommended to be coupled with genetic screening of family members.
Work over the past five years has identified additional genes that have implicated in causing familial TAAD. These include ROBO4 (roundabout guidance receptor 4), a gene that contributes to endothelial function [115], ARIH1 (Ariadne RBR E3 Ubiquitin Protein Ligase 1), a gene involved in nuclear localization and morphology [116], and two genes in the TGF-β signaling cascade, LTBP1 (latent TGF-β binding protein 1) and LTBP3 (latent TGF-β binding protein 3) [117][118]. Due to the fact that these are recent discoveries, more investigation needs to be done to determine the clinical outcomes of these patients.
3.6. Genetics of Sporadic TAAD
Recent work investigating the genetic risk factors of sporadic TAAD has generated further insights into this seemingly random disease that accounts for a large majority of cases of non-syndromic TAAD [7]. A cohort of 765 patients with sporadic TAAD had a GWAS peak at FBN1 even though patients did not present with any syndromic features of MFS, suggesting that even common variants in FBN1 can place patients at a higher risk for TAAD [20]. Novel variants in low-density lipoprotein receptor-related protein 1 (LRP1), a protein involved in endocytosis and intracellular signaling, and unc-51-like kinase 4 (ULK4), involved in endocytosis and axon growth, were also identified in another large cohort study, further suggesting that other genes may play a role [119]. A recent study used blood gene signatures to identify and validate four genes—CLU (Clusterin), DES (Desmin), MYH10, and FBLN5 (Fibulin 5)—related to sporadic TAA [120]. Another recent study found that 9.3% of sporadic TAAD patients had a pathogenic variant in genes associated with familial or syndromic TAAD (TGFBR, COL3A1, SMAD, ACTA2, TGFB2, or TGFBR2), indicating that genetic screening would benefit these patients and their families [79]. In addition, higher rates of variants of unknown significance (VUS) compared to control genomes suggests low yet stochastic penetrance and may be triggered by other physiologic factors such as hypertension [79]. Therefore, further research needs to be conducted on identifying non-syndromic mutations that place patients at higher risk for TAAD along with investigating screening strategies to detect these individuals [121].
References
- Kuzmik, G.A.; Sang, A.X.; Elefteriades, J.A. Natural history of thoracic aortic aneurysms. J. Vasc. Surg. 2012, 56, 565–571.
- Elefteriades, J.A.; Sang, A.; Kuzmik, G.; Hornick, M. Guilt by association: Paradigm for detecting a silent killer (thoracic aortic aneurysm). Open Heart 2015, 2, e000169.
- Web-based Injury Statistics Query and Reporting System (WISQARS). Available online: (accessed on 26 August 2019).
- Zhan, S.; Hong, S.; Shan-Shan, L.; Chen-Ling, Y.; Lai, W.; Dong-Wei, S.; Chao-Yang, T.; Xian-Hong, S.; Chun-Sheng, W. Misdiagnosis of aortic dissection: Experience of 361 patients. J. Clin. Hypertens. 2012, 14, 256–260.
- Zafar, M.A.; Farkas, E.A.; Javier, A.; Anderson, M.; Gilani, O.; Elefteriades, J.A. Are thromboembolic and bleeding complications a drawback for composite aortic root replacement? Ann. Thorac. Surg. 2012, 94, 737–743.
- Ziganshin, B.A.; Rajbanshi, B.G.; Tranquilli, M.; Fang, H.; Rizzo, J.A.; Elefteriades, J.A. Straight deep hypothermic circulatory arrest for cerebral protection during aortic arch surgery: Safe and effective. J. Thorac. Cardiovasc. Surg. 2014, 148, 888–898.
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606.
- Albornoz, G.; Coady, M.A.; Roberts, M.; Davies, R.R.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Familial thoracic aortic aneurysms and dissections--incidence, modes of inheritance, and phenotypic patterns. Ann. Thorac. Surg. 2006, 82, 1400–1405.
- Brownstein, A.J.; Kostiuk, V.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: 2018 Update and Clinical Implications. Aorta 2018, 6, 13–20.
- Keramati, A.R.; Sadeghpour, A.; Farahani, M.M.; Chandok, G.; Mani, A. The non-syndromic familial thoracic aortic aneurysms and dissections maps to 15q21 locus. BMC Med. Genet. 2010, 11, 143.
- Teixidó-Tura, G.; Franken, R.; Galuppo, V.; Gutiérrez García-Moreno, L.; Borregan, M.; Mulder, B.J.; García-Dorado, D.; Evangelista, A. Heterogeneity of aortic disease severity in patients with Loeys-Dietz syndrome. Heart 2016, 102, 626–632.
- Lindsay, M.E.; Dietz, H.C. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 2011, 473, 308–316.
- Faivre, L.; Collod-Beroud, G.; Adès, L.; Arbustini, E.; Child, A.; Callewaert, B.L.; Loeys, B.; Binquet, C.; Gautier, E.; Mayer, K.; et al. The new Ghent criteria for Marfan syndrome: What do they change? Clin. Genet. 2012, 81, 433–442.
- Iams, H.D. Diagnosis and management of Marfan syndrome. Curr. Sports Med. Rep. 2010, 9, 93–98.
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010, 121, e266–e369.
- Milewicz, D.M.; Michael, K.; Fisher, N.; Coselli, J.S.; Markello, T.; Biddinger, A. Fibrillin-1 (FBN1) mutations in patients with thoracic aortic aneurysms. Circulation 1996, 94, 2708–2711.
- Biddinger, A.; Rocklin, M.; Coselli, J.; Milewicz, D.M. Familial thoracic aortic dilatations and dissections: A case control study. J. Vasc. Surg. 1997, 25, 506–511.
- Coady, M.A.; Davies, R.R.; Roberts, M.; Goldstein, L.J.; Rogalski, M.J.; Rizzo, J.A.; Hammond, G.L.; Kopf, G.S.; Elefteriades, J.A. Familial patterns of thoracic aortic aneurysms. Arch. Surg. 1999, 134, 361–367.
- Hasham, S.N.; Lewin, M.R.; Tran, V.T.; Pannu, H.; Muilenburg, A.; Willing, M.; Milewicz, D.M. Nonsyndromic genetic predisposition to aortic dissection: A newly recognized, diagnosable, and preventable occurrence in families. Ann. Emerg. Med. 2004, 43, 79–82.
- LeMaire, S.A.; McDonald, M.L.; Guo, D.C.; Russell, L.; Miller, C.C.; Johnson, R.J.; Bekheirnia, M.R.; Franco, L.M.; Nguyen, M.; Pyeritz, R.E.; et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat. Genet. 2011, 43, 996–1000.
- Mimler, T.; Nebert, C.; Eichmair, E.; Winter, B.; Aschacher, T.; Stelzmueller, M.E.; Andreas, M.; Ehrlich, M.; Laufer, G.; Messner, B. Extracellular matrix in ascending aortic aneurysms and dissections—What we learn from decellularization and scanning electron microscopy. PLoS ONE 2019, 14, e0213794.
- Milewicz, D.M.; Trybus, K.M.; Guo, D.C.; Sweeney, H.L.; Regalado, E.; Kamm, K.; Stull, J.T. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 26–34.
- Wheeler, J.B.; Ikonomidis, J.S.; Jones, J.A. Connective tissue disorders and cardiovascular complications: The indomitable role of transforming growth factor-beta signaling. Adv. Exp. Med. Biol. 2014, 802, 107–127.
- Eckersley, A.; Mellody, K.T.; Pilkington, S.; Griffiths, C.E.M.; Watson, R.E.B.; O’Cualain, R.; Baldock, C.; Knight, D.; Sherratt, M.J. Structural and compositional diversity of fibrillin microfibrils in human tissues. J. Biol. Chem. 2018, 293, 5117–5133.
- Sakai, L.Y.; Keene, D.R.; Engvall, E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J. Cell. Biol. 1986, 103, 2499–2509.
- Davis, M.R.; Summers, K.M. Structure and function of the mammalian fibrillin gene family: Implications for human connective tissue diseases. Mol. Genet. Metab. 2012, 107, 635–647.
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339.
- Arnaud, P.; Hanna, N.; Aubart, M.; Leheup, B.; Dupuis-Girod, S.; Naudion, S.; Lacombe, D.; Milleron, O.; Odent, S.; Faivre, L.; et al. Homozygous and compound heterozygous mutations in the FBN1 gene: Unexpected findings in molecular diagnosis of Marfan syndrome. J. Med. Genet. 2017, 54, 100–103.
- Ziganshin, B.A.; Bailey, A.E.; Coons, C.; Dykas, D.; Charilaou, P.; Tanriverdi, L.H.; Liu, L.; Tranquilli, M.; Bale, A.E.; Elefteriades, J.A. Routine Genetic Testing for Thoracic Aortic Aneurysm and Dissection in a Clinical Setting. Ann. Thorac. Surg. 2015, 100, 1604–1611.
- Elefteriades, J.A.; Brownstein, A.J.; Ziganshin, B.A. Clinical and molecular genetics of aortic dissection. In Aortic Dissection Patients: True Stories and the Innovations that Saved Their Lives; Melissano, G., Chiesa, R., Eds.; Edi. Ermes: Milano, Italy, 2016; pp. 49–80. ISBN1 978-88-7051-565-7. (hardcover); ISBN2 978-88-7051-566-4. (digital edition).
- Pyeritz, R.E. The Marfan syndrome. Annu. Rev. Med. 2000, 51, 481–510.
- Judge, D.P.; Biery, N.J.; Keene, D.R.; Geubtner, J.; Myers, L.; Huso, D.L.; Sakai, L.Y.; Dietz, H.C. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Investig. 2004, 114, 172–181.
- Seo, G.H.; Kim, Y.M.; Kang, E.; Kim, G.H.; Seo, E.J.; Lee, B.H.; Choi, J.H.; Yoo, H.W. The phenotypic heterogeneity of patients with Marfan-related disorders and their variant spectrums. Medicine 2018, 97, e10767.
- Franken, R.; Groenink, M.; de Waard, V.; Feenstra, H.M.; Scholte, A.J.; van den Berg, M.P.; Pals, G.; Zwinderman, A.H.; Timmermans, J.; Mulder, B.J. Genotype impacts survival in Marfan syndrome. Eur. Heart J. 2016, 37, 3285–3290.
- Vollbrandt, T.; Tiedemann, K.; El-Hallous, E.; Lin, G.; Brinckmann, J.; John, H.; Bätge, B.; Notbohm, H.; Reinhardt, D.P. Consequences of cysteine mutations in calcium-binding epidermal growth factor modules of fibrillin-1. J. Biol. Chem. 2004, 279, 32924–32931.
- Faivre, L.; Collod-Beroud, G.; Loeys, B.L.; Child, A.; Binquet, C.; Gautier, E.; Callewaert, B.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M.; et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: An international study. Am. J. Hum. Genet. 2007, 81, 454–466.
- Reinhardt, D.P.; Mechling, D.E.; Boswell, B.A.; Keene, D.R.; Sakai, L.Y.; Bächinger, H.P. Calcium determines the shape of fibrillin. J. Biol. Chem. 1997, 272, 7368–7373.
- Tan, L.; Li, Z.; Zhou, C.; Cao, Y.; Zhang, L.; Li, X.; Cianflone, K.; Wang, Y.; Wang, D.W. FBN1 mutations largely contribute to sporadic non-syndromic aortic dissection. Hum. Mol. Genet. 2017, 26, 4814–4822.
- Chen, J.Z.; Sawada, H.; Moorleghen, J.J.; Weiland, M.; Daugherty, A.; Sheppard, M.B. Aortic Strain Correlates with Elastin Fragmentation in Fibrillin-1 Hypomorphic Mice. Circ. Rep. 2019, 1, 199–205.
- Kaartinen, V.; Warburton, D. Fibrillin controls TGF-beta activation. Nat. Genet. 2003, 33, 331–332.
- Jones, K.B.; Myers, L.; Judge, D.P.; Kirby, P.A.; Dietz, H.C.; Sponseller, P.D. Toward an understanding of dural ectasia: A light microscopy study in a murine model of Marfan syndrome. Spine 2005, 30, 291–293.
- Takeda, N.; Hara, H.; Fujiwara, T.; Kanaya, T.; Maemura, S.; Komuro, I. TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. Int. J. Mol. Sci. 2018, 19, 2125.
- Weinsaft, J.W.; Devereux, R.B.; Preiss, L.R.; Feher, A.; Roman, M.J.; Basson, C.T.; Geevarghese, A.; Ravekes, W.; Dietz, H.C.; Holmes, K.; et al. Aortic Dissection in Patients With Genetically Mediated Aneurysms: Incidence and Predictors in the GenTAC Registry. J. Am. Coll. Cardiol. 2016, 67, 2744–2754.
- Saeyeldin, A.; Zafar, M.A.; Velasquez, C.A.; Ip, K.; Gryaznov, A.; Brownstein, A.J.; Li, Y.; Rizzo, J.A.; Erben, Y.; Ziganshin, B.A.; et al. Natural history of aortic root aneurysms in Marfan syndrome. Ann. Cardiothorac. Surg. 2017, 6, 625–632.
- Roman, M.J.; Pugh, N.L.; Hendershot, T.P.; Devereux, R.B.; Dietz, H.; Holmes, K.; Eagle, K.A.; LeMaire, S.A.; Milewicz, D.M.; Morris, S.A.; et al. Aortic Complications Associated With Pregnancy in Marfan Syndrome: The NHLBI National Registry of Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions (GenTAC). J. Am. Heart Assoc. 2016, 5, e004052.
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798.
- Van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126.
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927.
- MacCarrick, G.; Black, J.H.; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587.
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell. Biol. 2012, 13, 616–630.
- Daugherty, A.; Chen, Z.; Sawada, H.; Rateri, D.L.; Sheppard, M.B. Transforming Growth Factor-β in Thoracic Aortic Aneurysms: Good, Bad, or Irrelevant? J. Am. Heart Assoc. 2017, 6, e00521.
- Loeys, B.L.; Chen, J.; Neptune, E.R.; Judge, D.P.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275–281.
- Akhurst, R.J. The paradoxical TGF-β vasculopathies. Nat. Genet. 2012, 44, 838–839.
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634.
- Germain, D.P. Ehlers-Danlos syndrome type IV. Orphanet J. Rare Dis. 2007, 2, 32.
- Byers, P. Vascular Ehlers-Danlos Syndrome. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2019.
- Carter, J.; Fenves, A.Z. Understanding vascular-type Ehlers-Danlos syndrome and avoiding vascular complications. Bayl. Univ. Med. Cent. Proc. 2017, 30, 52–53.
- Legrand, A.; Devriese, M.; Dupuis-Girod, S.; Simian, C.; Venisse, A.; Mazzella, J.M.; Auribault, K.; Adham, S.; Frank, M.; Albuisson, J.; et al. Frequency of de novo variants and parental mosaicism in vascular Ehlers-Danlos syndrome. Genet. Med. 2019, 21, 1568–1575.
- Shalhub, S.; Black, J.H.; Cecchi, A.C.; Xu, Z.; Griswold, B.F.; Safi, H.J.; Milewicz, D.M.; McDonnell, N.B. Molecular diagnosis in vascular Ehlers-Danlos syndrome predicts pattern of arterial involvement and outcomes. J. Vasc. Surg. 2014, 60, 160–169.
- Cortini, F.; Marinelli, B.; Seia, M.; De Giorgio, B.; Pesatori, A.C.; Montano, N.; Bassotti, A. Next-generation sequencing and a novel COL3A1 mutation associated with vascular Ehlers-Danlos syndrome with severe intestinal involvement: A case report. J Med. Case Rep. 2016, 10, 303.
- Schievink, W.I. Cerebrovascular Involvement in Ehlers-Danlos Syndrome. Curr. Treat. Options Cardiovasc. Med. 2004, 6, 231–236.
- North, K.N.; Whiteman, D.A.; Pepin, M.G.; Byers, P.H. Cerebrovascular complications in Ehlers-Danlos syndrome type IV. Ann. Neurol. 1995, 38, 960–964.
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888.
- Persikov, A.V.; Pillitteri, R.J.; Amin, P.; Schwarze, U.; Byers, P.H.; Brodsky, B. Stability related bias in residues replacing glycines within the collagen triple helix (Gly-Xaa-Yaa) in inherited connective tissue disorders. Hum. Mutat. 2004, 24, 330–337.
- Schwarze, U.; Schievink, W.I.; Petty, E.; Jaff, M.R.; Babovic-Vuksanovic, D.; Cherry, K.J.; Pepin, M.; Byers, P.H. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am. J. Hum. Genet. 2001, 69, 989–1001.
- Liu, X.; Wu, H.; Byrne, M.; Krane, S.; Jaenisch, R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 1852–1856.
- Prockop, D.J.; Kivirikko, K.I. Heritable diseases of collagen. N. Engl. J. Med. 1984, 311, 376–386.
- El Masri, H.; Loong, T.H.; Meurette, G.; Podevin, J.; Zinzindohoue, F.; Lehur, P.A. Bowel perforation in type IV vascular Ehlers-Danlos syndrome. A systematic review. Tech. Coloproctol. 2018, 22, 333–341.
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.M.; Bal-Theoleyre, L.; Fauret, A.L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur. J. Hum. Genet. 2015, 23, 1657–1664.
- Leistritz, D.F.; Pepin, M.G.; Schwarze, U.; Byers, P.H. COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet. Med. 2011, 13, 717–722.
- Erbel, R.; Aboyans, V.; Boileau, C.; Bossone, E.; Bartolomeo, R.D.; Eggebrecht, H.; Evangelista, A.; Falk, V.; Frank, H.; Gaemperli, O.; et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2873–2926.
- Meester, J.A.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 2017, 19, 386–395.
- Huang, J.; Davis, E.C.; Chapman, S.L.; Budatha, M.; Marmorstein, L.Y.; Word, R.A.; Yanagisawa, H. Fibulin-4 deficiency results in ascending aortic aneurysms: A potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ. Res. 2010, 106, 583–592.
- Igoucheva, O.; Alexeev, V.; Halabi, C.M.; Adams, S.M.; Stoilov, I.; Sasaki, T.; Arita, M.; Donahue, A.; Mecham, R.P.; Birk, D.E.; et al. Fibulin-4 E57K Knock-in Mice Recapitulate Cutaneous, Vascular and Skeletal Defects of Recessive Cutis Laxa 1B with both Elastic Fiber and Collagen Fibril Abnormalities. J. Biol. Chem. 2015, 290, 21443–21459.
- Callewaert, B.; Renard, M.; Hucthagowder, V.; Albrecht, B.; Hausser, I.; Blair, E.; Dias, C.; Albino, A.; Wachi, H.; Sato, F.; et al. New insights into the pathogenesis of autosomal-dominant cutis laxa with report of five ELN mutations. Hum. Mutat. 2011, 32, 445–455.
- Callewaert, B.L.; Willaert, A.; Kerstjens-Frederikse, W.S.; De Backer, J.; Devriendt, K.; Albrecht, B.; Ramos-Arroyo, M.A.; Doco-Fenzy, M.; Hennekam, R.C.; Pyeritz, R.E.; et al. Arterial tortuosity syndrome: Clinical and molecular findings in 12 newly identified families. Hum. Mutat. 2008, 29, 150–158.
- Ma, W.G.; Chou, A.S.; Mok, S.C.M.; Ziganshin, B.A.; Charilaou, P.; Zafar, M.A.; Sieller, R.S.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Positive family history of aortic dissection dramatically increases dissection risk in family members. Int. J. Cardiol. 2017, 240, 132–137.
- Elefteriades, J.A.; Farkas, E.A. Thoracic aortic aneurysm clinically pertinent controversies and uncertainties. J. Am. Coll. Cardiol. 2010, 55, 841–857.
- Guo, D.C.; Hostetler, E.M.; Fan, Y.; Kulmacz, R.J.; Zhang, D.; Nickerson, D.A.; Leal, S.M.; LeMaire, S.A.; Regalado, E.S.; Milewicz, D.M. Heritable Thoracic Aortic Disease Genes in Sporadic Aortic Dissection. J. Am. Coll. Cardiol. 2017, 70, 2728–2730.
- Luyckx, I.; Loeys, B.L. The genetic architecture of non-syndromic thoracic aortic aneurysm. Heart 2015, 101, 1678–1684.
- Sherrah, A.G.; Andvik, S.; van der Linde, D.; Davies, L.; Bannon, P.G.; Padang, R.; Vallely, M.P.; Wilson, M.K.; Keech, A.C.; Jeremy, R.W. Nonsyndromic Thoracic Aortic Aneurysm and Dissection: Outcomes With Marfan Syndrome Versus Bicuspid Aortic Valve Aneurysm. J. Am. Coll. Cardiol. 2016, 67, 618–626.
- Bradley, T.J.; Bowdin, S.C.; Morel, C.F.; Pyeritz, R.E. The Expanding Clinical Spectrum of Extracardiovascular and Cardiovascular Manifestations of Heritable Thoracic Aortic Aneurysm and Dissection. Can. J. Cardiol. 2016, 32, 86–99.
- Guo, D.C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493.
- Morisaki, H.; Akutsu, K.; Ogino, H.; Kondo, N.; Yamanaka, I.; Tsutsumi, Y.; Yoshimuta, T.; Okajima, T.; Matsuda, H.; Minatoya, K.; et al. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum. Mutat. 2009, 30, 1406–1411.
- Andelfinger, G.; Loeys, B.; Dietz, H. A Decade of Discovery in the Genetic Understanding of Thoracic Aortic Disease. Can. J. Cardiol. 2016, 32, 13–25.
- Ince, H.; Nienaber, C.A. Etiology, pathogenesis and management of thoracic aortic aneurysm. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 418–427.
- Karimi, A.; Milewicz, D.M. Structure of the Elastin-Contractile Units in the Thoracic Aorta and How Genes That Cause Thoracic Aortic Aneurysms and Dissections Disrupt This Structure. Can. J. Cardiol. 2016, 32, 26–34.
- Disabella, E.; Grasso, M.; Gambarin, F.I.; Narula, N.; Dore, R.; Favalli, V.; Serio, A.; Antoniazzi, E.; Mosconi, M.; Pasotti, M.; et al. Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle alpha-actin 2 (ACTA2). Heart 2011, 97, 321–326.
- Guo, D.C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 2009, 84, 617–627.
- Regalado, E.S.; Guo, D.C.; Prakash, S.; Bensend, T.A.; Flynn, K.; Estrera, A.; Safi, H.; Liang, D.; Hyland, J.; Child, A.; et al. Aortic Disease Presentation and Outcome Associated With ACTA2 Mutations. Circ. Cardiovasc. Genet. 2015, 8, 457–464.
- Renard, M.; Callewaert, B.; Baetens, M.; Campens, L.; MacDermot, K.; Fryns, J.P.; Bonduelle, M.; Dietz, H.C.; Gaspar, I.M.; Cavaco, D.; et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFβ signaling in FTAAD. Int. J. Cardiol. 2013, 165, 314–321.
- Milewicz, D.M.; Guo, D.C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic basis of thoracic aortic aneurysms and dissections: Focus on smooth muscle cell contractile dysfunction. Annu. Rev. Genomics Hum. Genet. 2008, 9, 283–302.
- Milewicz, D.M.; Ostergaard, J.R.; Ala-Kokko, L.M.; Khan, N.; Grange, D.K.; Mendoza-Londono, R.; Bradley, T.J.; Olney, A.H.; Ades, L.; Maher, J.F.; et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J. Med. Genet. A 2010, 152, 2437–2443.
- El-Hamamsy, I.; Yacoub, M.H. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat. Rev. Cardiol. 2009, 6, 771–786.
- Bellini, C.; Wang, S.; Milewicz, D.M.; Humphrey, J.D. Myh11(R247C/R247C) mutations increase thoracic aorta vulnerability to intramural damage despite a general biomechanical adaptivity. J. Biomech. 2015, 48, 113–121.
- Cheng, J.; Zhou, X.; Jiang, X.; Sun, T. Deletion of ACTA2 in mice promotes angiotensin II induced pathogenesis of thoracic aortic aneurysms and dissections. J. Thorac. Dis. 2018, 10, 4733–4740.
- Regalado, E.S.; Guo, D.C.; Estrera, A.L.; Buja, L.M.; Milewicz, D.M. Acute aortic dissections with pregnancy in women with ACTA2 mutations. Am J. Med. Genet. A 2014, 164, 106–112.
- Wang, L.; Guo, D.C.; Cao, J.; Gong, L.; Kamm, K.E.; Regalado, E.; Li, L.; Shete, S.; He, W.Q.; Zhu, M.S.; et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet. 2010, 87, 701–707.
- Stull, J.T.; Tansey, M.G.; Tang, D.C.; Word, R.A.; Kamm, K.E. Phosphorylation of myosin light chain kinase: a cellular mechanism for Ca2+ desensitization. Mol. Cell. Biochem. 1993, 127–128, 229–237.
- Gao, N.; Huang, J.; He, W.; Zhu, M.; Kamm, K.E.; Stull, J.T. Signaling through myosin light chain kinase in smooth muscles. J. Biol. Chem. 2013, 288, 7596–7605.
- Wallace, S.E.; Regalado, E.S.; Gong, L.; Janda, A.L.; Guo, D.C.; Russo, C.F.; Kulmacz, R.J.; Hanna, N.; Jondeau, G.; Boileau, C.; et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet. Med. 2019, 21, 144–151.
- Shalata, A.; Mahroom, M.; Milewicz, D.M.; Limin, G.; Kassum, F.; Badarna, K.; Tarabeih, N.; Assy, N.; Fell, R.; Cohen, H.; et al. Fatal thoracic aortic aneurysm and dissection in a large family with a novel MYLK gene mutation: Delineation of the clinical phenotype. Orphanet J. Rare Dis. 2018, 13, 41.
- Zhu, L.; Vranckx, R.; Khau Van Kien, P.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.M.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349.
- Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Scherer, S.; Liu, Y.; Presley, C.; Guo, D.; Estrera, A.L.; Safi, H.J.; Brasier, A.R.; et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 2007, 16, 2453–2462.
- Harakalova, M.; van der Smagt, J.; de Kovel, C.G.; Van’t Slot, R.; Poot, M.; Nijman, I.J.; Medic, J.; Joziasse, I.; Deckers, J.; Roos-Hesselink, J.W.; et al. Incomplete segregation of MYH11 variants with thoracic aortic aneurysms and dissections and patent ductus arteriosus. Eur. J. Hum. Genet. 2013, 21, 487–493.
- He, R.; Guo, D.C.; Estrera, A.L.; Safi, H.J.; Huynh, T.T.; Yin, Z.; Cao, S.N.; Lin, J.; Kurian, T.; Buja, L.M.; et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J. Thorac. Cardiovasc. Surg. 2006, 131, 671–678.
- Francis, S.H.; Corbin, J.D. Cyclic nucleotide-dependent protein kinases: Intracellular receptors for cAMP and cGMP action. Crit. Rev. Clin. Lab. Sci. 1999, 36, 275–328.
- Guo, D.C.; Regalado, E.; Casteel, D.E.; Santos-Cortez, R.L.; Gong, L.; Kim, J.J.; Dyack, S.; Horne, S.G.; Chang, G.; Jondeau, G.; et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am. J. Hum. Genet. 2013, 93, 398–404.
- Zhang, W.; Han, Q.; Liu, Z.; Zhou, W.; Cao, Q. Exome sequencing reveals a de novo PRKG1 mutation in a sporadic patient with aortic dissection. BMC Med. Genet. 2018, 19, 218.
- Kuang, S.Q.; Medina-Martinez, O.; Guo, D.C.; Gong, L.; Regalado, E.S.; Reynolds, C.L.; Boileau, C.; Jondeau, G.; Prakash, S.K.; Kwartler, C.S.; et al. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J. Clin. Investig. 2016, 126, 948–961.
- Lee, V.S.; Halabi, C.M.; Hoffman, E.P.; Carmichael, N.; Leshchiner, I.; Lian, C.G.; Bierhals, A.J.; Vuzman, D.; Mecham, R.P.; Frank, N.Y.; et al. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc. Natl. Acad. Sci. USA 2016, 113, 8759–8764.
- Guo, D.C.; Gong, L.; Regalado, E.S.; Santos-Cortez, R.L.; Zhao, R.; Cai, B.; Veeraraghavan, S.; Prakash, S.K.; Johnson, R.J.; Muilenburg, A.; et al. MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am. J. Hum. Genet. 2015, 96, 170–177.
- Combs, M.D.; Knutsen, R.H.; Broekelmann, T.J.; Toennies, H.M.; Brett, T.J.; Miller, C.A.; Kober, D.L.; Craft, C.S.; Atkinson, J.J.; Shipley, J.M.; et al. Microfibril-associated glycoprotein 2 (MAGP2) loss of function has pleiotropic effects in vivo. J. Biol. Chem. 2013, 288, 28869–28880.
- Koenig, S.N.; LaHaye, S.; Feller, J.D.; Rowland, P.; Hor, K.N.; Trask, A.J.; Janssen, P.M.; Radtke, F.; Lilly, B.; Garg, V. Notch1 haploinsufficiency causes ascending aortic aneurysms in mice. JCI Insight 2017, 2, 91353.
- Gould, R.A.; Aziz, H.; Woods, C.E.; Seman-Senderos, M.A.; Sparks, E.; Preuss, C.; Wünnemann, F.; Bedja, D.; Moats, C.R.; McClymont, S.A.; et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat. Genet. 2019, 51, 42–50.
- Tan, K.L.; Haelterman, N.A.; Kwartler, C.S.; Regalado, E.S.; Lee, P.T.; Nagarkar-Jaiswal, S.; Guo, D.C.; Duraine, L.; Wangler, M.F.; Bamshad, M.J.; et al. Ari-1 Regulates Myonuclear Organization Together with Parkin and Is Associated with Aortic Aneurysms. Dev. Cell 2018, 45, 226–244.
- Quiñones-Pérez, B.; VanNoy, G.E.; Towne, M.C.; Shen, Y.; Singh, M.N.; Agrawal, P.B.; Smith, S.E. Three-generation family with novel contiguous gene deletion on chromosome 2p22 associated with thoracic aortic aneurysm syndrome. Am J. Med. Genet. A 2018, 176, 560–569.
- Guo, D.C.; Regalado, E.S.; Pinard, A.; Chen, J.; Lee, K.; Rigelsky, C.; Zilberberg, L.; Hostetler, E.M.; Aldred, M.; Wallace, S.E.; et al. LTBP3 Pathogenic Variants Predispose Individuals to Thoracic Aortic Aneurysms and Dissections. Am. J. Hum. Genet. 2018, 102, 706–712.
- Guo, D.C.; Grove, M.L.; Prakash, S.K.; Eriksson, P.; Hostetler, E.M.; LeMaire, S.A.; Body, S.C.; Shalhub, S.; Estrera, A.L.; Safi, H.J.; et al. Genetic Variants in LRP1 and ULK4 Are Associated with Acute Aortic Dissections. Am. J. Hum. Genet. 2016, 99, 762–769.
- Xu, H.; Chen, S.; Zhang, H.; Zou, Y.; Zhao, J.; Yu, J.; Le, S.; Cui, J.; Jiang, L.; Wu, J.; et al. Network-based analysis reveals novel gene signatures in the peripheral blood of patients with sporadic nonsyndromic thoracic aortic aneurysm. J. Cell. Physiol. 2020, 235, 2478–2491.
- Kwartler, C.S.; Gong, L.; Chen, J.; Wang, S.; Kulmacz, R.; Duan, X.Y.; Janda, A.; Huang, J.; Kamm, K.E.; Stull, J.T.; et al. Variants of Unknown Significance in Genes Associated with Heritable Thoracic Aortic Disease Can Be Low Penetrant Risk Variants. Am. J. Hum. Genet. 2018, 103, 138–143.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
28 Jun 2021
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No