 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Olga Shilova | + 2386 word(s) | 2386 | 2021-05-18 08:40:56 | | | |
| 2 | Lindsay Dong | -9 word(s) | 2377 | 2021-06-28 10:14:52 | | | | |
| 3 | Lindsay Dong | -9 word(s) | 2377 | 2021-06-28 10:16:20 | | |
Video Upload Options
Cancer cells frequently overexpress specific surface receptors providing tumor growth and survival which can be used for precise therapy. Targeting cancer cell receptors with protein toxins is an attractive approach widely used in contemporary experimental oncology and preclinical studies.
1. Introduction
Cancer treatment has traditionally been based on surgery, radiation, and chemotherapy, which have shown limited therapeutic benefits in patients with metastatic disease. Despite significant advances in the development of systemic treatment, traditional chemotherapeutic agents cause serious side toxicity, restricting treatment to certain therapeutic dosages. In light of this, new approaches to selective treatment are urgently needed.
Protein toxins possessing such features as high cytotoxicity and efficiency have become promising components for anticancer therapy. Cancer cells frequently upregulate surface receptors that promote growth and survival, that is why various antigen-specific proteins including antibodies, antibody fragments (e.g., Fab and scFv), and other protein scaffolds (e.g., affibody and DARPin) have been developed as a moiety to target cancer cells [1][2]. Being genetically encoded, toxins can be expressed as fusion proteins with targeting moieties mentioned above and can have a wide range of modifications to prolong circulation in the bloodstream and increase tumor retention. Complete biodegradation within an organism is also an important advantage of protein toxins as anticancer agents [3][4].
In addition to natural protein toxins, designed toxins are also used in experimental oncology, for example, as an alternative to chemical photosensitizers [5][6][7][8]. The main advantage of protein photosensitizers is the opportunity to use a genetic engineering approach to combine cytotoxic and targeting moieties, avoiding chemical conjugation.
2. Soluble Targeted Toxins
2.1. Targeting and Toxic Modules Coupling Strategies
The history of targeted toxins began with the chemical conjugation of natural diphtheria toxin (DT) with anti-lymphocyte antibodies or their F(ab)2 fragments to produce agents for killing lymphoblastoid tumor cells [9]. This strategy helped to couple cell-specific delivery of antibodies with extremely high toxicity of DT, previously shown for mammalian cells [10]. The first generation of immunotoxins used chemical conjugation to couple natural toxins with full-length antibodies [11]. The introduction of hybridoma technology [12] enabled the production of precisely characterized bifunctional agents with a certain specificity. The second generation of immunotoxins arise due to the use of truncated fragments of protein toxins, lacking natural tropism, which helped to reduce in vivo side toxicity [13]. Over time, the variety of toxins used in the design of targeted therapy has grown [14][15], but the next breakthrough was made due to molecular cloning, which allowed for the production of the third-generation immunotoxins: fusion proteins consisting of antibody fragments linked to enzymatically active toxin domains [16][17].
2.2. Factors Affecting a Targeted Toxin Efficiency
Soluble targeted toxins are thought to be the embodiment of a “magic bullet” idea. Being applied systemically, these agents can reach disseminated, metastatic, or inoperable tumors and kill cancer cells. Still, there are several factors affecting the efficiency of targeted toxins (summarized in Figure 1).
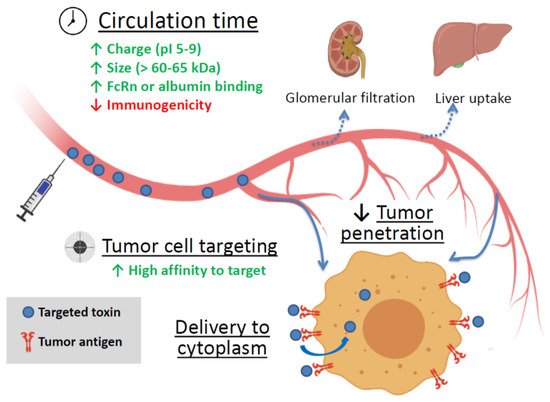
Figure 1. The main factors affecting the efficiency of targeted toxin. Green up arrows—factors enhancing circulation time and tumor cell targeting. Red down arrow—reducing factor. FcRn is the neonatal Fc receptor.
3. Targeted Toxins as Components of Nanoagents
Despite the successful use of immunotoxins, immunotherapy strategies are still expensive, mainly due to the complicated preparation process. Immunotoxins can also stimulate the host immune system and trigger the production of neutralizing antibodies. Intravenous administration of targeted protein toxins may be characterized by poor pharmacokinetic profiles in addition to non-specific distribution in tissues and organs of the body and can cause serious side effects including systemic toxicity. Besides, the penetration of anticancer drugs into tumor tissues is usually low and the high doses of drugs are required for treatment [18][19]. The use of nanocarriers, especially the targeted ones, for delivering toxins to tumor foci may improve the pharmacokinetics and pharmacodynamics of agents, control drug release, improve the specificity, increase internalization and intracellular delivery, and reduce systemic toxicity [20]. Nanocarriers can facilitate selective accumulation in tumors via the enhanced permeability and retention (EPR) effect and active cellular uptake [21]. Among various nanoscale drug carriers, liposomes, polymeric nanoparticles and noble metal nanoparticles have demonstrated the greatest potential in clinical application [22][23][24].
Liposomes have proven to be an efficient vehicle for delivering a high molecular weight neurotoxin botulinum toxin A to treat hypersensitive bladder and overactive bladder (OAB) without systemic injection [25].
Recently, a new method has been proposed for the preparation of small (80–90 nm) unilamellar antigen-targeted liposomes containing large amounts (thousands of protein molecules per liposome) of highly toxic PE40 [26] (Figure 2a). Efficient encapsulation of the proteins was achieved through electrostatic interaction between positively charged toxin proteins at pH lower than pI and negatively charged liposome membrane. The external surface of proteoliposomes were functionalized with covalently coupled DARPin_9-29 using “click chemistry” through a relatively long flexible linker. Functionalized proteoliposomes specifically bind to HER2-positive cells and after internalization cause cell death at subnanomolar concentrations [27].
Figure 2. (a) Small unilamellar targeted liposomes containing PE40 [26]; (b) Hybrid biofunctional nanocomplex based on radioactive 90Y bearing core-shell UCNP and functionalized with targeted toxin DARPin-PE40 [28].
4. Cytotoxic Mechanisms of Natural Toxins
The killing mechanisms of protein toxins can vary, but they differ from the mechanisms that are implemented in conventional chemotherapy [4], so an obtained resistance to chemotherapeutic agents does not affect the effectiveness of protein toxins. Furthermore, the mechanism complementation can provide a synergistic effect of combined therapy. In addition, protein toxins are not mutagens and should not accelerate tumor progression due to enhanced mutagenesis. They can be mass-produced cheaply in bacteria as homogeneous proteins [16].
Toxins of bacterial and plant origin commonly used as cytotoxic component in chimeric proteins in anticancer therapy are summarized in Table 1. The most toxic proteins include enzymes that inhibit translation at the elongation step. Unsurprisingly, most of them arise from natural toxins that have been effectively preselected by evolution.
| Mechanism of Action | Details | Examples | References |
|---|---|---|---|
| eEF2 inactivation | ADP-ribosylates elongation factor 2 (eEF2) and halt protein synthesis at the elongation step | Pseudomonas exotoxin A (PE, ETA) | [29][30] |
| Diphtheria toxin (DT) | [9][31] | ||
| Ribosome inactivation | N-glycosidase depurinates a critical adenine in 28S rRNA, which results in the inability of the ribosome to bind elongation factor 2, thereby blocking protein translation | Ricin | [32][33][34] |
| Shiga toxin (Stx) | [35] | ||
| Abrin | [36][37][38] | ||
| RNA degradation | Nonspecific RNA cleavage blocks protein synthesis and leads to apoptosis | Barnase | [39][40] |
| Binase | [41] | ||
| Cell signaling disruption | The cleavages of the MAP kinase family members leading to their inactivation; uncontrolled conversion of ATP to cAMP | Anthrax toxin | [42] |
| Photoinduced ROS production | The proteins absorb exciting light and produce reactive oxygen species | KillerRed | [43][44] |
| miniSOG | [45] | ||
| Direct apoptosis induction | Effector caspases cleavage | Granzyme B | [46] |
| Enhanced diffusion of anticancer drug | Vascular network modulation | Botulinum neurotoxin | [47][48] |
| Pore formation for better intracellular delivery | Listeriolysin O | [49][50] | |
| Streptolysin-O | [51][52] |
5. Reducing Protein Toxins Side Toxicity
The protein toxins high toxicity is one of main advantages of these molecules but at the same time it increases the risk and severity of side effects. The side toxicity of a protein can be based on a direct cell killing and inflammation induction [53]. The most common side effects caused by DT, PE, and ricin include vascular leak syndrome, hepatotoxicity, and kidney damage [54][55][56]. In addition, Shiga toxin is notorious for its ability to cause hemolytic uremic syndrome (HUS), potentially leading to life-threatening complications [57][58]. The production of neutralizing antibodies can also serve as a cause on side effects due to anaphylaxis reactions.
To date the number of strategies were developed to reduce protein drug off-target toxicity, the main tools are summarized in Table 2.
| Strategy Used for Side Toxicity Reduction | Principle | References |
|---|---|---|
| Impairment of natural tropism | Removing the natural targeting domains of AB toxins | [59] |
| Introduction of point mutations attenuating the target binding | [60] | |
| Construction of miniaturized toxin variants | Deletion of protein parts not directly involved in toxin mechanism of action to reduce any non-specific interaction and immunogenicity | [61][62][63] |
| Tumor-specific activation of a toxin | The replacement of furin cleavage site to tumor-specific proteases cleavage sites (MMP, uPA) | [64][65][66] |
| RES cells inactivation | Macrophages blockade decreasing toxic nanoparticles uptake | [67][68] |
6. Conclusions
The use of several toxic mechanisms or several target molecules makes it possible to compensate for the deficiencies of effector molecules, increase their efficiency and avoid selection of resistant cells. The designed toxic proteins capable of ROS production and fused to UCNP or luciferase make it possible to overcome the shallow depth of excitation light penetration, thus providing a novel approach to PDT of deeply located tumors.
Despite the numerous breakthrough solutions in cancer treatment, the problem is still far from being solved. It is worth mentioning that only two toxin-based molecules, namely Diphtheria toxin-based DAB389IL2 and DAB389IL3 [89][90], have been approved in late-stage clinical evaluation. Recently, a PE-based immunotoxin Moxetumomab Pasudotox (Lumoxiti), targeting CD22, has been approved for the treatment of patients with hairy cell leukemia [62]. In the future, new targeted therapies and combinations with increased selective anticancer activity and minimal side effects will be studied, which will increase the clinical efficacy of patients with various types of cancer.
References
- Deyev, S.M.; Lebedenko, E.N. Modern Technologies for Creating Synthetic Antibodies for Clinical Application. Acta Nat. 2009, 1, 32–50.
- Shilova, O.N.; Deyev, S.M. DARPins: Promising scaffolds for theranostics. Acta Nat. 2019, 11, 42–53.
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671.
- Serna, N.; Sánchez-García, L.; Unzueta, U.; Díaz, R.; Vázquez, E.; Mangues, R.; Villaverde, A. Protein-Based Therapeutic Killing for Cancer Therapies. Trends Biotechnol. 2018, 36, 318–335.
- Bulina, M.E.; Chudakov, D.M.; Britanova, O.V.; Yanushevich, Y.G.; Staroverov, D.B.; Chepurnykh, T.V.; Merzlyak, E.M.; Shkrob, M.A.; Lukyanov, S.; Lukyanov, K.A. A genetically encoded photosensitizer. Nat. Biotechnol. 2006, 24, 95–99.
- Mironova, K.E.; Proshkina, G.M.; Ryabova, A.V.; Stremovskiy, O.A.; Lukyanov, S.A.; Petrov, R.V.; Deyev, S.M. Genetically encoded immunophotosensitizer 4D5scFv-miniSOG is a highly selective agent for targeted photokilling of tumor cells in vitro. Theranostics 2013, 3, 831–840.
- Proshkina, G.M.; Shilova, O.N.; Ryabova, A.V.; Stremovskiy, O.A.; Deyev, S.M. A new anticancer toxin based on HER2/neu-specific DARPin and photoactive flavoprotein miniSOG. Biochimie 2015, 118, 116–122.
- Sarkisyan, K.S.; Zlobovskaya, O.A.; Gorbachev, D.A.; Bozhanova, N.G.; Sharonov, G.V.; Staroverov, D.B.; Egorov, E.S.; Ryabova, A.V.; Solntsev, K.M.; Mishin, A.S.; et al. KillerOrange, a Genetically Encoded Photosensitizer Activated by Blue and Green Light. PLoS ONE 2015, 10, e0145287.
- Ross, W.C.J.; Thorpe, P.E.; Cumber, A.J.; Edwards, D.C.; Hinson, C.A.; Davies, A.J.S. Increased Toxicity of Diphtheria Toxin for Human Lymphoblastoid Cells following Covalent Linkage to Anti-(human lymphocyte) Globulin or Its F(ab′)2 Fragment. Eur. J. Biochem. 1980, 104, 381–390.
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment a introduced into a cell can kill the cell. Cell 1978, 15, 245–250.
- Antignani, A.; FitzGerald, D. Immunotoxins: The role of the toxin. Toxins (Basel) 2013, 5, 1486–1502.
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497.
- Blythman, H.E.; Casellas, P.; Gros, O.; Gros, P.; Jansen, F.K.; Paolucci, F.; Pau, B.; Vidal, H. Immunotoxins: Hybrid molecules of monoclonal antibodies and a toxin subunit specifically kill tumour cells. Nature 1981, 290, 145–146.
- Pastan, I.; Willingham, M.C.; FitzGerald, D.J.P. Immunotoxins. Cell 1986, 47, 641–648.
- Vitetta, E.S.; Uhr, J.W. Immunotoxins: Redirecting nature’s poisons. Cell 1985, 41, 653–654.
- Pastan, I.; FitzGerald, D. Recombinant toxins for cancer treatment. Science 1991, 254, 1173–1177.
- FitzGerald, D.; Pastan, I. Redirecting Pseudomonas exotoxin. Semin. Cell Biol. 1991, 2, 31–37.
- Tannock, I.F.; Lee, C.M.; Tunggal, J.K.; Cowan, D.S.M.; Egorin, M.J. Limited Penetration of Anticancer Drugs through Tumor Tissue. Clin. Cancer Res. 2002, 8, 878–884.
- Sokolova, E.; Kutova, O.; Grishina, A.; Pospelov, A.; Guryev, E.; Schulga, A.; Deyev, S.; Balalaeva, I. Penetration efficiency of antitumor agents in ovarian cancer spheroids: The case of recombinant targeted toxin DARPin-LoPE and the chemotherapy drug, doxorubicin. Pharmaceutics 2019, 11, 219.
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146.
- Tahmasbi Rad, A.; Chen, C.W.; Aresh, W.; Xia, Y.; Lai, P.S.; Nieh, M.P. Combinational Effects of Active Targeting, Shape, and Enhanced Permeability and Retention for Cancer Theranostic Nanocarriers. ACS Appl. Mater. Interfaces 2019, 11, 10505–10519.
- Yan, W.; Leung, S.S.Y.; To, K.K.W. Updates on the use of liposomes for active tumor targeting in cancer therapy. Nanomedicine 2019, 15, 303–318.
- Palazzolo, S.; Bayda, S.; Hadla, M.; Caligiuri, I.; Corona, G.; Toffoli, G.; Rizzolio, F. The Clinical Translation of Organic Nanomaterials for Cancer Therapy: A Focus on Polymeric Nanoparticles, Micelles, Liposomes and Exosomes. Curr. Med. Chem. 2017, 25, 4224–4268.
- van Elk, M.; Murphy, B.P.; Eufrásio-da-Silva, T.; O’Reilly, D.P.; Vermonden, T.; Hennink, P.W.E.; Duffy, G.P.; Ruiz-Hernández, E. Nanomedicines for advanced cancer treatments: Transitioning towards responsive systems. Int. J. Pharm. 2016, 515, 132–164.
- Janicki, J.; Chancellor, M.; Kaufman, J.; Gruber, M.; Chancellor, D. Potential Effect of Liposomes and Liposome-Encapsulated Botulinum Toxin and Tacrolimus in the Treatment of Bladder Dysfunction. Toxins 2016, 8, 81.
- Deyev, S.; Proshkina, G.; Baryshnikova, O.; Ryabova, A.; Avishai, G.; Katrivas, L.; Giannini, C.; Levi-Kalisman, Y.; Kotlyar, A. Selective staining and eradication of cancer cells by protein-carrying DARPin-functionalized liposomes. Eur. J. Pharm. Biopharm. 2018, 130, 296–305.
- Sokolova, E.; Proshkina, G.; Kutova, O.; Shilova, O.; Ryabova, A.; Schulga, A.; Stremovskiy, O.; Zdobnova, T.; Balalaeva, I.; Deyev, S. Recombinant targeted toxin based on HER2-specific DARPin possesses a strong selective cytotoxic effect in vitro and a potent antitumor activity in vivo. J. Control. Release 2016, 233, 48–56.
- Guryev, E.L.; Volodina, N.O.; Shilyagina, N.Y.; Gudkov, S.V.; Balalaeva, I.V.; Volovetskiy, A.B.; Lyubeshkin, A.V.; Sen’, A.V.; Ermilov, S.A.; Vodeneev, V.A.; et al. Radioactive (90Y) upconversion nanoparticles conjugated with recombinant targeted toxin for synergistic nanotheranostics of cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 9690–9695.
- Collier, R.J.; Gilliland, D.G.; Lory, S. Structure-activity relationships in diphtheria toxin and exotoxin A from Pseudomonas aeruginosa. Prog. Clin. Biol. Res. 1979, 31, 751–759.
- Kreitman, R.J.; Pastan, I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011, 17, 6398–6405.
- Vingerhoeds, M.H.; Steerenberg, P.A.; Hendriks, J.J.G.W.; Crommelin, D.J.A.; Storm, G. Targeted delivery of diphtheria toxin via immunoliposomes: Efficient antitumor activity in the presence of inactivating anti-diphtheria toxin antibodies. FEBS Lett. 1996, 395, 245–250.
- Lord, J.M.; Roberts, L.M.; Robertus, J.D. Ricin: Structure, mode of action, and some current applications. FASEB J. 1994, 8, 201–208.
- Fodstad, Ø.; Kvalheim, G.; Godal, A.; Lotsberg, J.; Aamdal, S.; HOst, H.; Pihl, A. Phase I Study of the Plant Protein Ricin. Cancer Res. 1984, 44, 862–865.
- Li, C.; Yan, R.; Yang, Z.; Wang, H.; Zhang, R.; Chen, H.; Wang, J. BCMab1-Ra, a novel immunotoxin that BCMab1 antibody coupled to Ricin A chain, can eliminate bladder tumor. Oncotarget 2017, 8, 46704–46705.
- Engedal, N.; Skotland, T.; Torgersen, M.L.; Sandvig, K. Shiga toxin and its use in targeted cancer therapy and imaging. Microb. Biotechnol. 2011, 4, 32–46.
- Godal, A.; Fodstad, O.Y.; Pihl, A. Studies On The Mechanism Of Action Of Abrin-9.2.27 Immunotoxin In Human Melanoma Cell Lines. Cancer Res. 1987, 47, 6243–6247.
- Wawrzynczak, E.J.; Zangemeister-Wittke, U.; Waibel, R.; Henry, R.V.; Parnell, G.D.; Cumber, A.J.; Jones, M.; Stahel, R.A. Molecular and biological properties of an abrin a chain immunotoxin designed for therapy of human small cell lung cancer. Br. J. Cancer 1992, 66, 361–366.
- Griffiths, G.D.; Leek, M.D.; Gee, D.J. The toxic plant proteins ricin and abrin induce apoptotic changes in mammalian lymphoid tissues and intestine. J. Pathol. 1987, 151, 221–229.
- Edelweiss, E.; Balandin, T.G.; Ivanova, J.L.; Lutsenko, G.V.; Leonova, O.G.; Popenko, V.I.; Sapozhnikov, A.M.; Deyev, S.M. Barnase as a new therapeutic agent triggering apoptosis in human cancer cells. PLoS ONE 2008, 3.
- Balandin, T.G.; Edelweiss, E.; Andronova, N.V.; Treshalina, E.M.; Sapozhnikov, A.M.; Deyev, S.M. Antitumor activity and toxicity of anti-HER2 immunoRNase scFv 4D5-dibarnase in mice bearing human breast cancer xenografts. Invest. New Drugs 2011, 29, 22–32.
- Mironova, N.L.; Petrushanko, I.Y.; Patutina, O.A.; Sen’kova, A.V.; Simonenko, O.V.; Mitkevich, V.A.; Markov, O.V.; Zenkova, M.A.; Makarov, A.A. Ribonuclease binase inhibits primary tumor growth and metastases via apoptosis induction in tumor cells. Cell Cycle 2013, 12, 2120–2131.
- Bachran, C.; Leppla, S.H. Tumor targeting and drug delivery by anthrax toxin. Toxins 2016, 8, 197.
- Liang, L.; Lu, Y.; Zhang, R.; Care, A.; Ortega, T.A.; Deyev, S.M.; Qian, Y.; Zvyagin, A.V. Deep-penetrating photodynamic therapy with KillerRed mediated by upconversion nanoparticles. Acta Biomater. 2017, 51, 461–470.
- Yuan, M.; Liu, C.; Li, J.; Ma, W.; Yu, X.; Zhang, P.; Ji, Y. The effects of photodynamic therapy on leukemia cells mediated by KillerRed, a genetically encoded fluorescent protein photosensitizer. BMC Cancer 2019, 19.
- Souslova, E.A.; Mironova, K.E.; Deyev, S.M. Applications of genetically encoded photosensitizer miniSOG: From correlative light electron microscopy to immunophotosensitizing. J. Biophotonics 2017, 10, 338–352.
- Kurschus, F.C.; Jenne, D.E. Delivery and therapeutic potential of human granzyme B. Immunol. Rev. 2010, 235, 159–171.
- Ansiaux, R.; Gallez, B. Use of botulinum toxins in cancer therapy. Expert Opin. Investig. Drugs 2007, 16, 209–218.
- Jordan, B.F.; Gallez, B. Surrogate MR markers of response to chemo- or radiotherapy in association with co-treatments: A retrospective analysis of multi-modal studies. Contrast Media Mol. Imaging 2010, 5, 323–332.
- Pirie, C.M.; Liu, D.V.; Dane Wittrup, K. Targeted cytolysins synergistically potentiate cytoplasmic delivery of gelonin immunotoxin. Mol. Cancer Ther. 2013, 12, 1774–1782.
- Provoda, C.J.; Stier, E.M.; Lee, K.D. Tumor cell killing enabled by listeriolysin O-liposome-mediated delivery of the protein toxin gelonin. J. Biol. Chem. 2003, 278, 35102–35108.
- Walev, I.; Bhakdi, S.C.; Hofmann, F.; Djonder, N.; Valeva, A.; Aktories, K.; Bhakdi, S. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. USA 2001, 98, 3185–3190.
- Sakakibara, A.; Tsukuda, M.; Kondo, N.; Ishiguro, Y.; Kimura, M.; Fujita, K.; Takahashi, H.; Matsuda, H. Examination of the optimal condition on the in vitro sensitivity to telomelysin in head and neck cancer cell lines. Auris Nasus Larynx 2011, 38, 589–599.
- Alipour, M.; Pucaj, K.; Smith, M.G.; Suntres, Z.E. Toxicity of ricin toxin a chain in rats. Drug Chem. Toxicol. 2013, 36, 224–230.
- Pai, L.H.; Bookman, M.A.; Ozols, R.F.; Young, R.C.; Smith, J.W.; Longo, D.L.; Gould, B.; Frankel, A.; McClay, E.F.; Howell, S.; et al. Clinical evaluation of intraperitoneal Pseudomonas exotoxin immunoconjugate OVB3-PE in patients with ovarian cancer. J. Clin. Oncol. 1991, 9, 2095–2103.
- Shafiee, F.; Aucoin, M.G.; Jahanian-Najafabadi, A. Targeted Diphtheria Toxin-Based Therapy: A Review Article. Front. Microbiol. 2019, 10, 2340.
- Soler-Rodriguez, A.M.; Uhr, J.W.; Richardson, J.; Vitetta, E.S. The toxicity of chemically deglycosylated ricin A-chain in mice. Int. J. Immunopharmacol. 1992, 14, 281–291.
- Proulx, F.Ç.; Seidman, E.G.; Karpman, D. Pathogenesis of Shiga toxin—Associated hemolytic uremic syndrome. Pediatr. Res. 2001, 50, 163–171.
- Palermo, M.S.; Exeni, R.A.; Fernández, G.C. Hemolytic uremic syndrome: Pathogenesis and update of interventions. Expert Rev. Anti. Infect. Ther. 2009, 7, 697–707.
- Chen, K.H.; Liu, S.; Bankston, L.A.; Liddington, R.C.; Leppla, S.H. Selection of anthrax toxin protective antigen variants that discriminate between the cellular receptors TEM8 and CMG2 and achieve targeting of tumor cells. J. Biol. Chem. 2007, 282, 9834–9845.
- Mechaly, A.; McCluskey, A.J.; John Collier, R. Changing the receptor specificity of anthrax toxin. MBio 2012, 3, e00088-12.
- Onda, M.; Beers, R.; Xiang, L.; Lee, B.; Weldon, J.E.; Kreitman, R.J.; Pastan, I. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc. Natl. Acad. Sci. USA 2011, 108, 5742–5747.
- Mazor, R.; Pastan, I. Immunogenicity of Immunotoxins Containing Pseudomonas Exotoxin A: Causes, Consequences, and Mitigation. Front. Immunol. 2020, 11, 1261.
- Sokolova, E.A.; Shilova, O.N.; Kiseleva, D.V.; Schulga, A.A.; Balalaeva, I.V.; Deyev, S.M. HER2-Specific Targeted Toxin DARPin-LoPE: Immunogenicity and Antitumor Effect on Intraperitoneal Ovarian Cancer Xenograft Model. Int. J. Mol. Sci. 2019, 20, 2399.
- Liu, S.; Netzel-Arnett, S.; Birkedal-Hansen, H.; Leppla, S.H. Tumor Cell-selective Cytotoxicity of Matrix Metalloproteinase-activated Anthrax Toxin. Cancer Res. 2000, 60, 6061–6067.
- Liu, S.; Bugge, T.H.; Leppla, S.H. Targeting of Tumor Cells by Cell Surface Urokinase Plasminogen Activator-dependent Anthrax Toxin. J. Biol. Chem. 2001, 276, 17976–17984.
- Phillips, D.D.; Fattah, R.J.; Crown, D.; Zhang, Y.; Liu, S.; Moayeri, M.; Fischer, E.R.; Hansen, B.T.; Ghirlando, R.; Nestorovich, E.M.; et al. Engineering anthrax toxin variants that exclusively form octamers and their application to targeting tumors. J. Biol. Chem. 2013, 288, 9058–9065.
- Nikitin, M.P.; Zelepukin, I.V.; Shipunova, V.O.; Sokolov, I.L.; Deyev, S.M.; Nikitin, P.I. Enhancement of the blood-circulation time and performance of nanomedicines via the forced clearance of erythrocytes. Nat. Biomed. Eng. 2020, 4, 717–731.
- Mirkasymov, A.B.; Zelepukin, I.V.; Nikitin, P.I.; Nikitin, M.P.; Deyev, S.M. In vivo blockade of mononuclear phagocyte system with solid nanoparticles: Efficiency and affecting factors. J. Control. Release 2021, 330, 111–118.
- Kondo, T.; FitzGerald, D.; Chaudhary, V.K.; Adhya, S.; Pastan, I. Activity of immunotoxins constructed with modified Pseudomonas exotoxin A lacking the cell recognition domain. J. Biol. Chem. 1988, 263, 9470–9475.
- Batra, J.K.; Kasprzyk, P.G.; Bird, R.E.; Pastan, I.; King, C.R. Recombinant anti-erbB2 immunotoxins containing Pseudomonas exotoxin. Proc. Natl. Acad. Sci. USA 1992, 89, 5867–5871.
- Proshkina, G.M.; Kiseleva, D.V.; Shilova, O.N.; Ryabova, A.V.; Shramova, E.I.; Stremovskiy, O.A.; Deyev, S.M. Bifunctional Toxin DARP-LoPE Based on the Her2-Specific Innovative Module of a Non-Immunoglobulin Scaffold as a Promising Agent for Theranostics. Mol. Biol. 2017, 51, 865–873.
- Sokolova, E.A.; Stremovskiy, O.A.; Zdobnova, T.A.; Balalaeva, I.V.; Deyev, S.M. Recombinant immunotoxin 4D5scFv-PE40 for targeted therapy of HER2-positive tumors. Acta Nat. 2015, 7, 93–96.
- Hassan, R.; Sharon, E.; Thomas, A.; Zhang, J.; Ling, A.; Miettinen, M.; Kreitman, R.J.; Steinberg, S.M.; Hollevoet, K.; Pastan, I. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen. Cancer 2014, 120, 3311–3319.
- FitzGerald, D.J.; Wayne, A.S.; Kreitman, R.J.; Pastan, I. Treatment of hematologic malignancies with immunotoxins and antibody-drug conjugates. Cancer Res. 2011, 71, 6300–6309.
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149.
- Kreitman, R.J.; Stetler-Stevenson, M.; Margulies, I.; Noel, P.; FitzGerald, D.J.P.; Wilson, W.H.; Pastan, I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J. Clin. Oncol. 2009, 27, 2983–2990.
- Kreitman, R.J.; Pastan, I. Development of recombinant immunotoxins for hairy cell leukemia. Biomolecules 2020, 10, 1140.
- Weldon, J.E.; Xiang, L.; Chertov, O.; Margulies, I.; Kreitman, R.J.; Fitzgerald, D.J.; Pastan, I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 2009, 113, 3792–3800.
- Weldon, J.E.; Pastan, I. A guide to taming a toxin—Recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700.
- McCluskey, A.J.; Olive, A.J.; Starnbach, M.N.; Collier, R.J. Targeting HER2-positive cancer cells with receptor-redirected anthrax protective antigen. Mol. Oncol. 2013, 7, 440–451.
- Zahaf, N.I.; Lang, A.E.; Kaiser, L.; Fichter, C.D.; Lassmann, S.; McCluskey, A.; Augspach, A.; Aktories, K.; Schmidt, G. Targeted delivery of an ADP-ribosylating bacterial toxin into cancer cells. Sci. Rep. 2017, 7, 41252.
- Loftis, A.R.; Santos, M.S.; Truex, N.L.; Biancucci, M.; Satchell, K.J.F.; Pentelute, B.L. Anthrax Protective Antigen Retargeted with Single-Chain Variable Fragments Delivers Enzymes to Pancreatic Cancer Cells. ChemBioChem 2020, 21, 2772–2776.
- Gage, E.; Hernandez, M.O.; O’Hara, J.M.; McCarthy, E.A.; Mantis, N.J. Role of the mannose receptor (CD206) in innate immunity to ricin toxin. Toxins 2011, 3, 1131–1145.
- Simmons, B.M.; Stahl, P.D.; Russell, J.H. Mannose receptor-mediated uptake of ricin toxin and ricin A chain by macrophages. Multiple intracellular pathways for a chain translocation. J. Biol. Chem. 1986, 261, 7912–7920.
- Mantis, N.J.; Farrant, S.A.; Mehta, S. Oligosaccharide Side Chains on Human Secretory IgA Serve as Receptors for Ricin. J. Immunol. 2004, 172, 6838–6845.
- Thorpe, P.E.; Wallace, P.M.; Knowles, P.P.; Relf, M.G.; Brown, A.N.; Watson, G.J.; Blakey, D.C.; Newell, D.R. Improved Antitumor Effects of Immunotoxins Prepared with Deglycosylated Ricin A-Chain and Hindered Disulfide Linkages. Cancer Res. 1988, 48, 6396–6403.
- Blakey, D.C.; Watson, G.J.; Knowles, P.P.; Thorpe, P.E. Effect of chemical deglycosylation of ricin A chain on the in vivo fate and cytotoxic activity of an immunotoxin composed of ricin A chain and anti-Thy 1.1 antibody. Cancer Res. 1987, 47, 947–952.
- Liu, S.; Wang, H.; Currie, B.M.; Molinolo, A.; Leung, H.J.; Moayeri, M.; Basile, J.R.; Alfano, R.W.; Gutkind, J.S.; Frankel, A.E.; et al. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J. Biol. Chem. 2008, 283, 529–540.
- Allahyari, H.; Heidari, S.; Ghamgosha, M.; Saffarian, P.; Amani, J. Immunotoxin: A new tool for cancer therapy. Tumor Biol. 2017, 39, 1–11.
- Antignani, A.; Ho, E.C.H.; Bilotta, M.T.; Qiu, R.; Sarnvosky, R.; Fitzgerald, D.J. Targeting receptors on cancer cells with protein toxins. Biomolecules 2020, 10, 1331.



