 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Agata Zieba | + 1631 word(s) | 1631 | 2021-06-17 08:03:16 | | | |
| 2 | Amina Yu | -4 word(s) | 1627 | 2021-06-28 03:28:07 | | |
Video Upload Options
The fatty acid amide hydrolase enzyme (FAAH) belongs to the serine hydrolase superfamily. It is involved in the degradation of biologically active lipids. Enzyme inhibitors may exhibit analgesic, anti-inflammatory, anxiolytic, and antidepressant activity. Importantly, blockade of FAAH does not cause undesirable side effects of direct cannabinoid agonists. Due to that fact, its blockade became an emerging strategy in the treatment of several central nervous system (CNS) and peripheral diseases. The development of novel effective FAAH inhibitors became a key focus in drug design.
1. Introduction
The fatty acid amide hydrolase enzyme (FAAH) belongs to the serine hydrolase superfamily. It is involved in the degradation of biologically active lipids—endocannabinoids, e.g., anandamide and 2-arachidonoylglycerol—or related-amidated signaling lipids. FAAH activity is considered to play an essential role in the development of certain pathological conditions [1]. Enzyme inhibitors may exhibit analgesic, anti-inflammatory, anxiolytic, and antidepressant activity. Importantly, blockade of FAAH does not cause undesirable side effects of direct cannabinoid agonists[2]. Due to that fact, its blockade became an emerging strategy in the treatment of several central nervous system (CNS) and peripheral diseases [1][2][3][4]. The development of novel effective FAAH inhibitors became a key focus in drug design[4] [5].
Computation resources are widely used in the examination of various properties of medicinal compounds. Quantitative structure–activity relationship methods require building mathematical or computational models to find a significant correlation between the structure and the biological activity of certain groups of substances [6]. These approaches quantitatively correlate the relationships between the chemical structure alterations and respective changes in bioactivity. Their usage enables us to optimize the properties of currently used chemicals and predict various parameters that refer to the biological activity of untested and sometimes unavailable compounds [6].
2. QSAR studies performed on FAAH inhibitors
Unfortunately, there is minimal information about the structure–activity relationship studies performed on a series of different types of fatty acid amide hydrolase inhibitors[7] . Käsnänen et al. synthesized a series of meta-substituted phenolic N-alkyl/aryl carbamates. They later submitted these compounds into the 3D-QSAR analysis, which revealed some interesting structure-activity correlations, e.g., the presence of a meta-substituted phenyl ring positively influenced the biological activity of these compounds [8]. Zhao et al. constructed a pharmacophore model for FAAH antagonists based on 21 typical compounds available in the literature. It contained four essential features—two H-bond acceptor units, a hydrophobic part, and one aromatic ring unit. This model was successfully applied to the prediction of the activity of 55 compounds[9]. Later, Han et al. decided to evaluate a series of 26 novel oleoylethanolamide derivatives using the CoMFA method [10]. That provided information about the desirable and unfavorable modifications which can be applied to enhance or decrease the biological activity of these molecules, e.g., a long aliphatic carbon chain enables the molecule to reach a green region of desired steric interactions and increases its potency. The paper published by Lorca et al. described a 3D-QSAR study performed on a series of 90 reported FAAH inhibitors that shared a common structural pattern—pyrimidinyl-piperazine-carboxamide moiety. Structure–activity conclusions obtained from the contour map analysis contributed to the designing of new compounds that showed promising predicted activities [11]. Recently, Zięba et al. published a study performed with the use of QSAR. This study was conducted on a series of FAAH ligands containing 1,3,4-oxadiazol-2-one moiety, designed and biologically tested by Patel et al. [12][13].
3. 1,3,4-oxadiazol-2-one as FAAH inhibitors
Among the diverse scaffolds utilized for the development of FAAH inhibitors, 1,3,4-oxadiazol-2-one has gained recent attention in the development of serine hydrolase inhibitors, including FAAH [7][13][14][15].
QSAR studies performed on the series of 31 fatty acid amide hydrolase inhibitors, containing 1,3,4-oxadiazol-2-one moiety, led to the formation of the CoMFA (Comparative Molecular Field Analysis) and CoMSIA (Comparative Molecular Similarity Indices Analysis) models. The obtained models were characterized by good statistical parameters: CoMFA Q2 = 0.61, R2 = 0.98; CoMSIA Q2 = 0.64, R2 = 0.93. The CoMFA model field contributions were 54.1% and 45.9% for steric and electrostatic fields, respectively. In the CoMSIA model, electrostatic, steric, hydrogen bond donor, and hydrogen acceptor properties were equal to 34.6%, 23.9%, 23.4%, and 18.0%, respectively. These models were validated by applying the leave-one-out technique, the seven-element test set (CoMFA r2test-set = 0.91; CoMSIA r2test-set = 0.91), a progressive scrambling test, and external validation criteria developed by Golbraikh and Tropsha (CoMFA r20 = 0.98, k = 0.95; CoMSIA r20 = 0.98, k = 0.89) [12].
As the statistical significance of the obtained model was confirmed, the results of the CoMFA and CoMSIA field calculation were mapped onto the enzyme binding site. That provided information on the contribution of specific moieties to the biological activity of these analogs [12].
3.1. The CoMFA fields evaluation
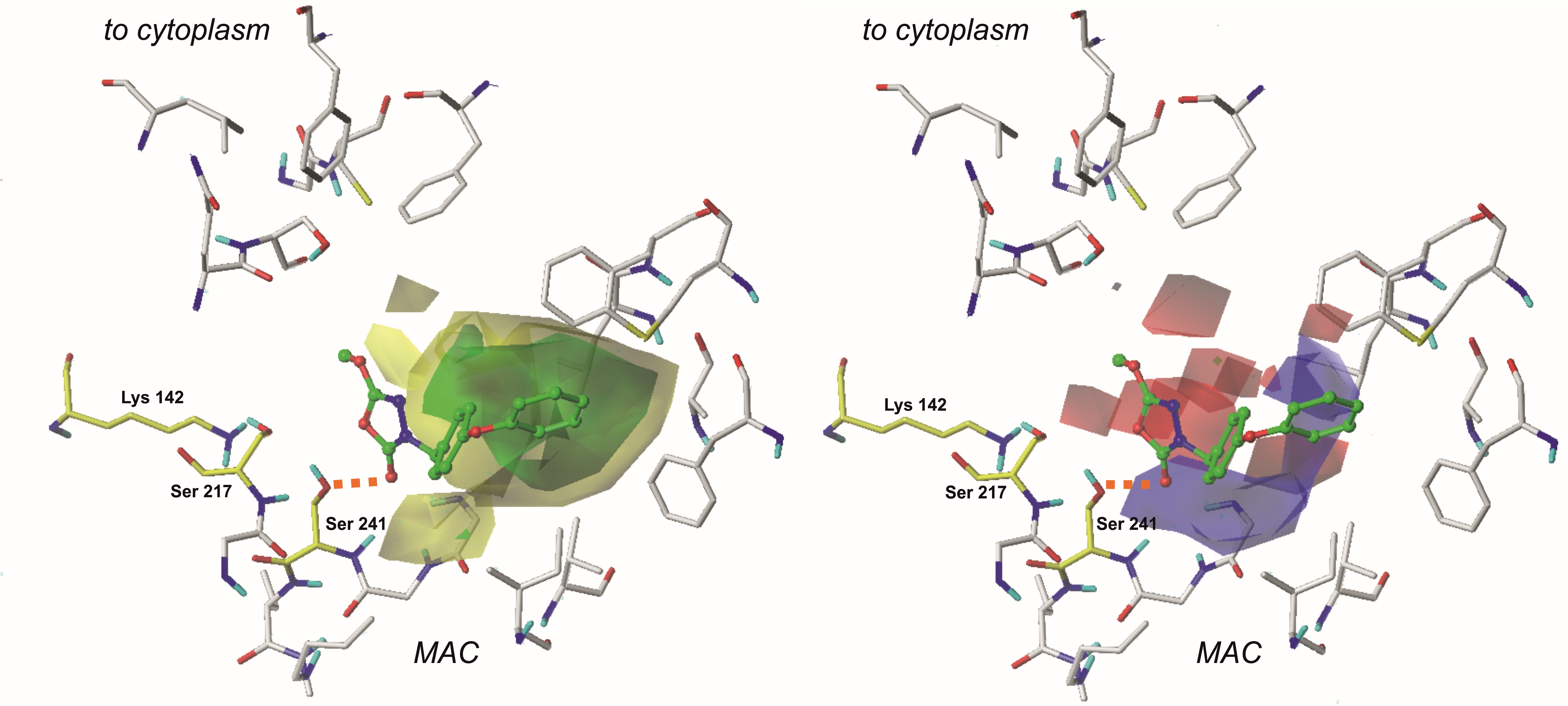

In particular, in the case of CoMFA fields, examination of electrostatic properties revealed red plots surrounding the 1,3,4-oxadiazol-2-one moiety. This highlights the importance of heterocycle, highly electronegative substituent in this area of each ligand. Due to that, compounds containing aromatic substituents in this position (characterized by more homogenous charge distribution) were less active.
Steric properties are visualized as green and yellow plots. The first one represents regions where bulky substituents are considered to influence the activity of the compound positively. Analysis of steric contour maps, viewed on the compounds from the training set, revealed that high-volume moieties, e.g., aromatic rings, are generally more desired on the opposite side to the 1,3,4-oxadiazol-2-one moiety. Thus, providing these regions with an additional 5- or 6- membered ring might positively influence the binding affinity. It is worth noting that molecules containing small-volume substituents attached to the 3rd position of the mentioned earlier heterocycle do not reach the green region of favorable interactions and are characterized by a lower biological activity.
3.2. The CoMSIA fields evaluation
Steric and electrostatic plots obtained in the CoMSIA modeling are in good agreement with the generated CoMFA fields. However, their contributions were relatively smaller than in the previously performed examination. In the CoMSIA study, two additional contour plots are generated—hydrogen bond donor and hydrogen bond acceptor.
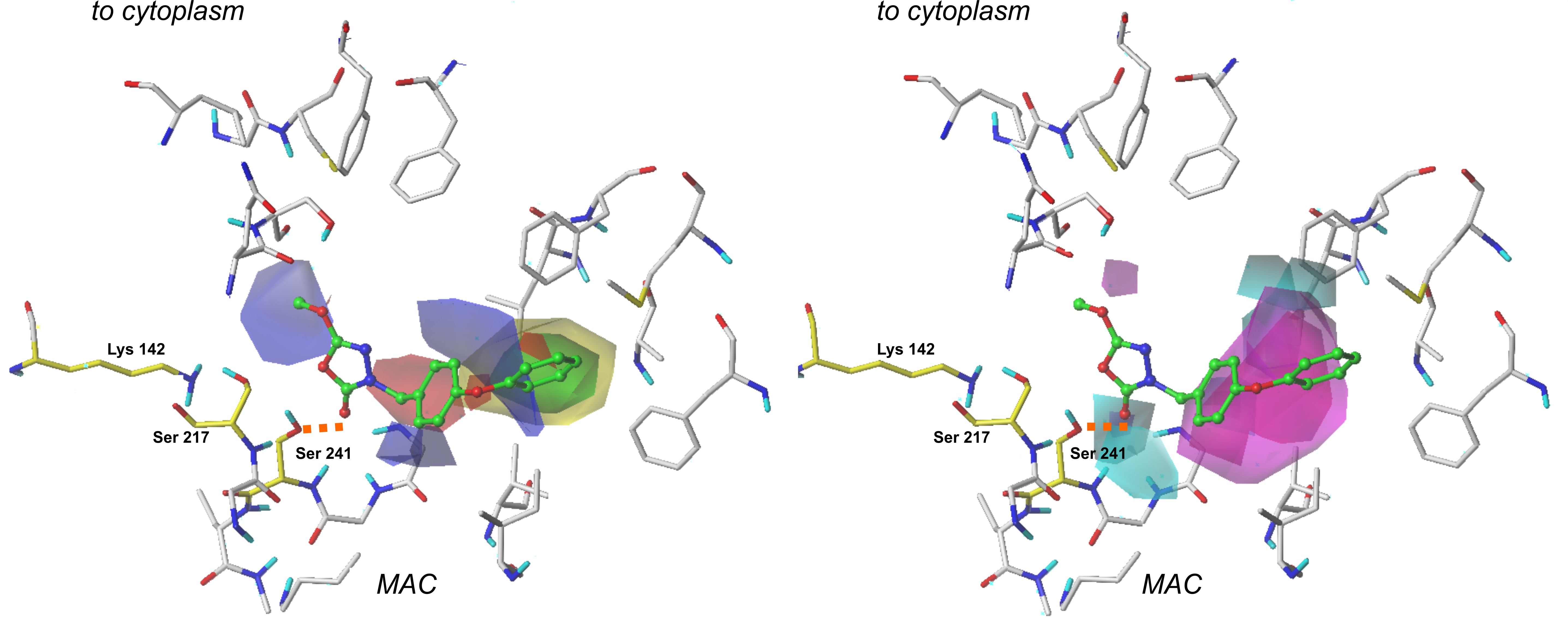 Figure 2. The CoMSIA Fields projected on the most active compound from the training set. The steric interactions (left part) are presented as yellow and green plots, while electrostatic properties (right part) are represented by red and blue fields [12].
Figure 2. The CoMSIA Fields projected on the most active compound from the training set. The steric interactions (left part) are presented as yellow and green plots, while electrostatic properties (right part) are represented by red and blue fields [12].
Hydrogen bond donor fields are represented by cyan and purple plots. In this context, cyan fields constitute favorable interactions, which means that hydrogen-bond-donating groups in these regions positively affect the molecule’s binding affinity. Purple marks indicate plots where H-bond donors are generally not required, and their presence results in the compound’s biological activity decrease.
A large plot showing favorable H-bond donor properties is located in the neighborhood of a terminal aromatic ring in our reference compounds. Hence, providing these structures with another amine or hydroxyl group might benefit the ligand’s binding affinity. It appears that another contour of this type is present close to the 1,3,4-oxadiazol-2-one ring and oxygen atom from the enzyme’s Ser 241 residue. This suggests that the examined area is essential for a binding of a ligand. Moreover, it is believed that introducing another hydrogen-bond acceptor group might positively influence binding affinity and the compound’s biological activity. Interestingly, the same oxygen atom from the carbonyl group (1,2,4-oxadiazol-2-one moiety) of a ligand is involved in other H-bond contacts with Ile 238 and Gly 239. These residues, along with Gly 240, form an oxyanion hole—a characteristic structure responsible for the stabilization of intermediates derived in the enzyme’s activation process. Introducing another hydrogen bond donor in this particular place may positively impact the compound’s biological activity.
When studying H-bond acceptor properties, magenta isopleths represent regions where the presence of hydrogen-bond-accepting groups will increase the biological activity of a compound. In contrast, such groups are not desired in fields encompassed by red plots. In the case of compound 1, a bulky isopleth indicating H-bond acceptor properties is located in a terminal’s phenoxy group neighborhood, which means that generally speaking, H-bond acceptors are desired in this area. Thus, providing the structure with more electronegative atoms that can act as H-bond acceptors will positively influence the ligand’s binding affinity [12].
4. Conclusions
References
- Piomelli, D. Endocannabinoids. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 194–196. ISBN 978-0-12-378631-9.
- Tripathi, R.K.P. A Perspective Review on Fatty Acid Amide Hydrolase (FAAH) Inhibitors as Potential Therapeutic Agents. Eur. J. Med. Chem. 2020, 188, 111953.
- Leweke, F.M.; Piomelli, D.; Pahlisch, F.; Muhl, D.; Gerth, C.W.; Hoyer, C.; Klosterkötter, J.; Hellmich, M.; Koethe, D. Cannabidiol Enhances Anandamide Signaling and Alleviates Psychotic Symptoms of Schizophrenia. Transl. Psychiatry 2012, 2, e94.
- Gaetani, S.; Dipasquale, P.; Romano, A.; Righetti, L.; Cassano, T.; Piomelli, D.; Cuomo, V. Chapter 5 The Endocannabinoid System as A Target for Novel Anxiolytic and Antidepressant Drugs. In International Review of Neurobiology; Academic Press: Amsterdam, The Netherlands, 2009; Volume 85, pp. 57–72.
- Mauro Mileni; Joie Garfunkle; Jessica DeMartino; Benjamin F. Cravatt; Dale L. Boger; Raymond C. Stevens; Binding and Inactivation Mechanism of a Humanized Fatty Acid Amide Hydrolase by α-Ketoheterocycle Inhibitors Revealed from Cocrystal Structures. Journal of the American Chemical Society 2009, 131, 10497-10506, 10.1021/ja902694n.
- Roy, K. . Advances in QSAR Modeling: Applications in Pharmaceutical, Chemical, Food, Agricultural and Environmental Sciences; Springer: New York, NY, USA, 2017; pp. ISBN 978-3-31-986018-3.
- Beliaev, A.; Ferreira, H.S.; Learmonth, D.A.; Bonifácio, M.J.; Torrão, L.; Pires, N.M.; Soares-da-Silva, P.; Kiss, L.E.; Synthesis and Structure–Activity Relationships of Ionizable 1,3,4-Oxadiazol-2(3H)-Ones as Peripherally Selective FAAH Inhibitors with Improved Aqueous Solubility. Pure Appl. Chem. 2016, 88, 341-347.
- Käsnänen, H.; Myllymäki, M.J.; Minkkilä, A.; Kataja, A.O.; Saario, S.M.; Nevalainen, T.; Koskinen, A.M.P.; Poso, A.; 3-Heterocycle-Phenyl N-Alkylcarbamates as FAAH Inhibitors: Design, Synthesis and 3D-QSAR Studies. ChemMedChem 2010, 5, 213-231.
- Xin Zhao; Minsheng Chen; Biyun Huang; Hong Ji; Mu Yuan; Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) Studies on α1A-Adrenergic Receptor Antagonists Based on Pharmacophore Molecular Alignment. International Journal of Molecular Sciences 2011, 12, 7022-7037, 10.3390/ijms12107022.
- Daxiong Han; Biyan Wang; Hui Jin; Haiyan Wang; Meimei Chen; Design, synthesis and CoMFA studies of OEA derivatives as FAAH inhibitors. Medicinal Chemistry Research 2017, 26, 2951-2966, 10.1007/s00044-017-1995-6.
- Marcos Lorca; Yudisladys Valdes; Hery Chung; Javier Romero-Parra; David Pessoa-Mahana; Jaime Mella; Romero- Parra; Pessoa- Mahana; Three-Dimensional Quantitative Structure-Activity Relationships (3D-QSAR) on a Series of Piperazine-Carboxamides Fatty Acid Amide Hydrolase (FAAH) Inhibitors as a Useful Tool for the Design of New Cannabinoid Ligands.. International Journal of Molecular Sciences 2019, 20, 2510, 10.3390/ijms20102510.
- Patel, J.Z.; Parkkari, T.; Laitinen, T.; Kaczor, A.A.; Saario, S.M.; Savinainen, J.R.; Navia-Paldanius, D.; Cipriano, M.; Leppänen, J.; Koshevoy, I.O.; et al. Chiral 1,3,4-Oxadiazol-2-Ones as Highly Selective FAAH Inhibitors. J. Med. Chem. 2013, 56, 8484–8496.
- Patel, J.Z.; van Bruchem, J.; Laitinen, T.; Kaczor, A.A.; Navia-Paldanius, D.; Parkkari, T.; Savinainen, J.R.; Laitinen, J.T.; Nevalainen, T.J. Revisiting 1,3,4-Oxadiazol-2-Ones: Utilization in the Development of ABHD6 Inhibitors. Bioorg. Med. Chem. 2015, 23, 6335–6345.
- Use of an Inhibitor to Identify Members of the Hormone-Sensitive Lipase Family. Biochemistry. Available online: https://pubs.acs.org/doi/abs/10.1021/bi0613978 (accessed on 9 March 2021).
- Muccioli, G.G.; Labar, G.; Lambert, D.M. CAY10499, a Novel Monoglyceride Lipase Inhibitor Evidenced by an Expeditious MGL Assay. ChemBioChem 2008, 9, 2704–2710.

