 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Paula Montero | + 2858 word(s) | 2858 | 2021-06-16 08:54:11 | | | |
| 2 | Amina Yu | Meta information modification | 2858 | 2021-06-25 11:57:18 | | |
Video Upload Options
Janus kinases (JAK) are a group of intracellular tyrosine kinases (JAK1, JAK2, JAK3, TYK2) that are crucial in signal transduction initiated by a wide range of membrane receptors. Signal transducer and activator of transcription (STAT) comprises a family of 7 members (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, STAT6), which function as transcription factors. The JAK/STAT signaling pathway controls multiple cellular processes that are essential for cell homeostasis.Between the different JAK/STAT isoforms, it appears that JAK2/STAT3 are predominant, initiating cellular changes observed in ILDs.
1. Introduction
Interstitial lung diseases (ILDs) comprise a heterogeneous group of disorders characterized by lung damage as a result of lung fibroblast proliferation, interstitial inflammation, and fibrosis [1]. Drug and radiation-induced lung disorders, pulmonary hemorrhages syndrome, chronic hypersensitivity pneumonitis, pulmonary sarcoidosis, or ILDs associated with autoimmune diseases like systemic sclerosis (SS) and rheumatoid arthritis (RA) are classified as interstitial lung diseases with known etiology [2]. The American Thoracic Society (ATS) and European Respiratory Society (ERS) subdivides the unknown etiology ILDs into three groups in which idiopathic interstitial pneumonias are the major ones [3][4]. Inside the major group (Table 1), there are chronic fibrosing disorders, such as idiopathic pulmonary fibrosis (IPF) and idiopathic nonspecific interstitial pneumonia (NSIP), smoking-related disorders, such as respiratory bronchiolitis–interstitial lung disease (RB–ILD), desquamative interstitial pneumonia (DIP), and acute/subacute idiopathic interstitial pneumonias, such as cryptogenic organizing pneumonia (COP) and acute interstitial pneumonia (AIP) [5].
| Major Idiopathic Interstitial Pneumonias | Histologic Findings | Radiologic Pattern |
|---|---|---|
| Idiopathic pulmonary fibrosis (IPF) | Heterogeneous areas of patchy lung fibrosis and UIP. [2] | Basal and peripheral reticular opacities with honeycombing and traction bronchiectasis. [6] |
| Idiopathic nonspecific interstitial pneumonia (NSIP) | Symmetric and homogeneous UIP. | Patchy ground-glass opacities and scattered micronodules. [7] |
| Respiratory bronchiolitis–interstitial lung disease (RB–ILD) | Alveolar macrophages within the bronchioles. | Centrilobular nodules. Central and peripheral bronchial wall thickening. [8] |
| Desquamative interstitial pneumonia (DIP) | Alveolar spaces with macrophages and desquamated alveolar cells. | Extensive and diffuse ground-glass opacities with peripheral and lower lobe predominance. [2] |
| Cryptogenic organizing pneumonia (COP) | Tissue polyps within the alveolar ducts and alveoli, with preservation of the lung architecture. | Patchy peripheral or peribronchial consolidations predominant in the lower lung lobes and multiple nodules. [9] |
| Acute interstitial pneumonia (AIP) | Diffuse alveolar damage. | Extensive ground-glass opacities and areas of consolidation. [10] |
In inflammation disease, the histology is characterized by organizing pneumonia or non-specific interstitial pneumonia, while fibrosis dominant disease is characterized by the usual interstitial pneumonia (UIP) pattern, characterized by fibroblastic foci and mild to moderate inflammation [11]. However, managing ILDs is a challenge for clinicians. Pathologic and clinical similarities amongst ILDs make diagnostic difficult. Therefore, multidisciplinary evaluation is usually recommended, including the discussion of clinical, radiological, and pathological data from lung biopsy if required [12].
that are crucial in signal transduction initiated by a wide range of membrane receptors [13]. Signal transducer and activator of transcription (STAT) comprises a family of 7 members (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, STAT6), which function as transcription factors [14]. The JAK/STAT signaling pathway controls multiple cellular processes that are essential for cell homeostasis. ILDs caused by COVID-19 infection, their relationship with JAK/STAT, and therapeutic strategies will be analyzed.
2. JAK/STAT Activation Mechanisms
Then, receptor-associated JAKs are activated and phosphorylate the tyrosine residues of the intracellular tail of their receptors. These phosphorylations serve as docking sites for signal transducers and activators of transcription (STATs) and bind to them through their SH2 domain. Thus, the STATs become activated by JAKs and translocate to the nucleus to regulate gene expression. This can happen whenever a receptor dimerizes in the absence of the ligand, when STATs dimers in the absence of the activating phosphorylation by a conformational change, or when latent STATs remain in a state of nuclear import and export [
Several cytokines, which activate a JAK/STAT pathway, like IL-4, IL-13, IL-6, IL-11, and IL-31, are implicated in the pathogenicity of ILDs.
IL-4 and IL-13 are related to ILDs [15] and are required for the maintenance of pulmonary fibrosis [16], modulating the abnormal activation of lung fibroblast [17]. Both cytokines share their receptor structure and common downstream signaling. From the two types of receptors, subunit 1 binds to TYK2, activating JAK1, STAT3, and STAT6 [18]. In AECs from both a BLM-induced fibrosis mouse model and pulmonary fibrosis patients,
IL-6 mediates different inflammatory processes in the lungs, and its dysregulated expression was implicated in the pathogenesis of interstitial pneumonias and IPF [19][20]. In a BLM-induced mouse model, IPF fibroblasts, and normal lung fibroblasts, STAT3 is activated after IL-6 binding to the receptor Stimulation of lung fibroblasts with IL-11 induces STAT3 phosphorylation [21]. Regarding gp130 activation by IL-31, there is not much evidence of its relationship with ILDs, but one study in a mouse pulmonary fibrosis model found that IL-31 signals through STAT1 activation, and may constitute a possible pathway of mouse pulmonary fibrosis [22].
JAK/STAT axis is also activated by growth factors. One of them is transforming growth factor β1 (TGF-β1), the principal pro-fibrotic factor promoting fibroblasts to myofibroblasts differentiation in ILDs [23]. Usually, its activation leads to the phosphorylation of the signal transducer proteins SMAD2/3, which translocate to the nucleus to activate pro-fibrotic genes. Other studies in human fibroblasts and mouse models of systemic sclerosis describe JAK2 activation by TGF-β1, which in turn phosphorylates STAT3 and leads to its nuclear translocation [24].
Moreover, p-JAK2 was found in the nucleus of fibrotic areas, a fact that implies that it may act independently of STAT3 [25]. The same signaling is observed in systemic sclerosis (SS), where TGF-β1 induces JAK2 phosphorylation, which then interacts with phosphorylated STAT3 to induce fibrotic responses. Interestingly, JAK2 may not only be a downstream target of TGF-β1 in fibroblasts but also performs positive feedback, amplifying TGF-β1 signaling by stimulating more TGF-β1 expression [26].
The JAK/STAT pathway and its different activators are widely described in cancer, but its implication in ILDs is still poorly understood. Therefore, some studies analyze the implication of JAK/STAT activation molecules in ILDs, but do not describe the mechanism, nor which JAKs and STATs are implicated in that activation. This happens with epidermal growth factor (EGF), platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and vascular endothelial growth factor (VEGF).
PDGF is increased in alveolar macrophages in patients with ILD [27] and bronchoalveolar lavage fluid (BALF) and lung samples from patients with IPF [28]. PDGF activates STAT1 signaling in primary lung fibroblasts [29] and JAK2/STAT3 in atrial fibroblasts in dogs with heart failure [30]. Recent evidence shows that VEGF constitutes an exhaled biomarker in IPF, with significant correlations with the clinical manifestations [31], and patients with idiopathic interstitial pneumonia have higher VEGF levels in plasma [32]. VEGF is associated with STAT3 in primary astrocytes when ionizing radiation-induced overexpression of VEGF leads to STAT3 activation [33].
3. Cellular Processes Activated by JAK/STAT
Figure 2shows the different cellular processes implicated in the pathogenesis of ILDs, and their relationship with JAK/STAT signaling pathway, which will be discussed below.
Fibroblast to mesenchymal transition (FMT) is a cellular process in which fibroblasts lose their differentiation and acquire the mesenchymal phenotype of myofibroblasts, characterized by the expression of alpha smooth muscle actin (α-SMA) and an aberrant expression of extracellular matrix components (ECM). Further, STAT3 regulates IL-6–mediated and TGF-β1–mediated myofibroblast differentiation in murine lung fibroblasts [34], and inhibition of STAT3 was found to reduce TGFβ1-induced FMT in fibroblasts, and ameliorate skin fibrosis in two mouse models of systemic sclerosis [35]. I secretion by lung fibroblasts in IPF and that enhanced expression of STAT3 in IPF fibroblasts might be responsible for their fibrogenic phenotype [36]. Consistent with these findings, our group observed that both p-STAT3 p-JAK2 inhibition in lung fibroblasts from IPF patients partially reduced FMT induced by TGF-β1 and IL-6/IL-13 [25].
Another cellular transformation found in fibrotic diseases is epithelial to mesenchymal transition (EMT), which allows epithelial cells to convert into motile mesenchymal cells [37]. This transition is implicated in the regulation of metastasis and cancer progression and is driven by the IL-6/JAK2/STAT3 pathway in lung and ovarian carcinomas [38][39]. Moreover, IL-6 secreted by activated fibroblasts in cancer stroma induces EMT of gastric cancer cells via JAK2/STAT3 In peritoneal fibrosis, IL-6/JAK2/STAT3 acts as a pro-EMT pathway [40].
In pulmonary fibrosis, EMT was described as a source of myofibroblasts in in vitro studies, animal models, and patients [41]. In IPF, EMT in ATII cells contributes to the pathology of the disease [42][43]. Nonetheless, there are no studies that describe STAT3 as a mediator of the EMT process in ILDs, although our group observed that TGF-β1 induced EMT in ATII cells was attenuated by dual p-JAK2/p- STAT3 inhibition, showing that this regulator might be directly involved [25].
Cellular senescence is a stable cell cycle arrest that maintains cells viable and metabolically active. This process is implicated in IPF pathogenesis. In cancer, IL-6 and STAT3 are related to the induction of senescence [44] and in lung fibroblasts, STAT3 is an important factor driving oxidant-induced senescence [45]. Waters and colleagues showed that targeting STAT3, protected against lung fibroblast cell senescence, similarly to our results, in which the senescence responses induced by TGF-β1 in fibroblasts and A549 alveolar type II cells were suppressed by dual si-RNA-JAK2/STAT3 inhibition [25].
As a result of senescence, apoptosis and proliferation are reduced [46]. However, opposite results are described in both cell processes related to ILDs.
Apoptosis is a process of programmed cell death that regulates cell number to maintain the homeostasis of many adult tissues. Moreover, STAT3 overexpression in human lung fibroblasts induces resistance to FasL-induced apoptosis [47]. Moodley et al. could associate the implication of STAT3 in IPF-fibroblast resistance to apoptosis, demonstrating that treatment of IPF fibroblasts with IL-6 conferred resistance to FasL-induced apoptosis, an effect mediated by STAT3 In lung fibroblasts derived from IPF patients, stimulation with IL-6 induced proliferation, and a transient STAT3 activation, while in normal fibroblasts, IL-6 stimulation reduced proliferation through STAT3 signaling [48].
The ER is regulated by different elements including protein load, cell metabolism, redox balance, and calcium homeostasis. When the protein folding capacity of the ER is altered, the unfolded protein response (UPR) activates to restore homeostasis. While there is evidence that ER stress drives pathogenesis pathways in ILDs, there are no studies on the role of JAK/STAT. Overall, these findings indicate the implication of STAT3 in ER stress, but how STAT3 is activated in response to ER stress specifically in ILDs needs further investigation.
Autophagy is a process in which the cell sequestrates dysfunctional cellular cytoplasmic components into vesicles to deliver them to the lysosome for destruction in order to maintain cell survival [49]. Normal fibroblasts activate autophagy in response to stress caused by collagen accumulation. In AECs from IPF patients, there is insufficient autophagy, which may lead to epithelial cell senescence, whereas in fibroblasts, lowered autophagy may induce differentiation into myofibroblasts [50]. Further, in BALF cells derived from patients with IPF and RA-ILD, gene expression analysis of autophagy markers showed similar expressions, which indicates that autophagy might be impaired in the same way as IPF in other ILDs [51].
In cancer, STAT3 activates genes of anti-autophagy proteins like BCL2, BCL2L1, and MCL1 [52], which disrupt the formation of the complex BECN1/PIK3C3, essential for autophagy [44]. Both nuclear and cytoplasmic STAT3s were found to contribute to the signaling of autophagy [53]. While the direct contribution of STAT in autophagy in ILDs has not been described, our group found that pharmacological inhibition of JAK2/STAT3 increased autophagy in the lungs of BLM-induced fibrosis in rats [25], which evidences a possible role for this pathway in autophagy in ILDs. However, more studies are still necessary to clarify this role.
4. JAK/STAT and COVID-19
After infection, ATII cells and alveolar macrophages trigger an inflammatory response that releases cytokines and chemokines and induces the recruitment of natural killer cells (NK), T-lymphocytes, blood-borne macrophages, and neutrophils, which aggravate the inflammatory response causing the cytokine storm [54]. This storm is characterized by the secretion of acute phase response cytokines IL-6, TNF-α, IL-1β, and IL-6 induces endothelial activation, inflammatory cell migration and macrophage activation, which in turn aggravates the inflammatory damage [55]. Acute respiratory distress syndrome (ARDS) can occur as a result of the aforementioned acute systemic inflammatory response [56].
The inflammation stage of COVID-19 shares some similarities with interstitial pneumonia seen in patients with ILD. In fact, it has already been reported that ILD is developed as an important consequence after COVID-19 infection [57][58]. The most common radiological pattern in COVID-19 is bilateral ground-glass opacification with or without consolidation in a subpleural distribution [59]. The histologic findings include diffuse alveolar damage (DAD), desquamation, interstitial fibrosis, and microcystic honeycombing.
COVID-19 patients also show fibrotic manifestations as a result of lung injury [60][61], and some of the molecular markers seen in COVID-19, are increased in IPF patients, which implies that pulmonary fibrosis has a role in the development of COVID-19 [62]. Indeed, it can be speculated that pulmonary injuries caused by COVID-19 in severe patients might progress to pulmonary fibrosis in the future [63]. As the pandemic is still in course, and not all data are available, we can rely on results from previous coronavirus outbreaks, like severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS), to predict how COVID-19-induced fibrosis will affect life quality of disease survivors. Some follow-up studies concluded that fibrosis appears as a common sequelae in severe SARS patients [64][65][66] and in a follow-up study of 36 patients surviving MERS, 33% had pulmonary fibrosis [67].
Due to the critical role of inflammation in the pathogenesis of COVID-19, and the histological and radiological similarities with ILDs, we will analyze the possible implication of JAK/STAT signaling in COVID-19 (Figure 1). Finally, another bioinformatic study has analyzed associated pathways of COVID-19 by exploring genetic profiles and making correlations between ACE2 and genes related to the JAK-STAT pathway in airway epithelial cells. However, JAK1, JAK3, STAT3, STAT5B, and STAT6 were found uncorrelated or negatively correlated [68], which is surprising due to the strong relationship of STAT3 in the pathogenesis of inflammatory and fibrotic diseases. Of note, more studies describing the molecular basis of the JAK/STAT pathway in COVID-19 are needed to address this question.
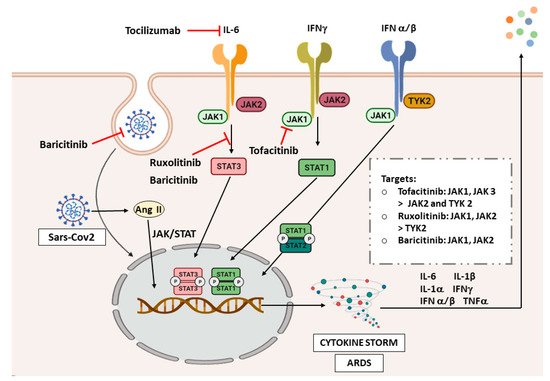
Figure 1. JAK/STAT inhibitors in COVID-19. Abbreviations: interferon (IFN), angiotensin II (Ang II), acute respiratory distress syndrome (ARDS). The image was created with BioRender.
COVID-19 has proven to cause inflammation and fibrosis that can be related to the findings in ILDs. Therefore, as the JAK/STAT pathway plays an important role in the pathogenesis of ILDs, and signals the inflammatory response in COVID-19, it is not surprising that one of the therapeutic strategies under investigation for COVID-19 is blocking JAK/STAT pathway. Figure 1 shows the different JAK/STAT inhibitors that are being studied and their targets in the signaling pathway. There are currently plenty of clinical trials assessing tocilizumab in COVID-19 patients, and it has shown promising efficacy in cases with respiratory failure and ARDS [69].
There is more evidence, though, on the use of ruxolitinib and baricitinib. The first randomized controlled trial on the use of ruxolitinib in patients with severe COVID-19 showed that patients receiving this drug plus standard-of-care had better improvement and safety than the control group [70]. One of them, a phase III multicenter study, was completed (NCT04362137), which assesses the efficacy and safety of ruxolitinib in patients with COVID-19 associated cytokine storm. Therefore, additional results from larger controlled studies are needed to confirm the possibility of a treatment benefit with ruxolitinib.
A phase II pilot study (NCT04358614) showed an improvement of clinical characteristics and respiratory function parameters in baricitinib-treated patients, although the sample size in this trial was very low [71]. There is also a completed phase III trial (NCT04401579) with no published results, which evaluates baricitinib and remdesivir in hospitalized adult patients. The safety profile of this treatment was proven in COVID-19 patients; however, more trials are needed to evaluate its effectiveness. To date, we retrieve 15 clinical trials analyzing baricitinib for the treatment of COVID-19.
The inflammatory process is a natural protective response when a virus attacks the body. The triggered cytokine release is primarily regulated by JAK/STAT signaling to activate the innate immunity response. It was shown that, in stages of the disease not requiring admittance to the hospital, nearly 80% of COVID-19 patients can clear the virus through this endogenous antiviral mechanism [72][73]. Therefore, is crucial to find the appropriate target patients for the use of these drugs [74], to not impair the natural viral clearance mechanisms, and patients with a stronger cytokine storm response, appear to benefit more from the treatment with JAK/STAT inhibitors [75].
Another important question to address is the management of fibrosis derived from COVID-19 infection. However, post-inflammatory pulmonary fibrosis in COVID-19 does not have a current treatment. However, ARDS is a risk factor for secondary pulmonary fibrosis in COVID-19 patients [62][76], and some of the clinical trials mentioned above for tofacitinib and ruxolitinib, were shown to ameliorate the cytokine storm and ARDS in COVID-19 patients. These effects show the therapeutic potential of JAK inhibitors in preventing the long-term fibrotic consequences by reducing lung injuries caused by ARDS and inflammation during COVID-19 and point them out as interesting strategies to reduce the risk factors, which will induce fibrosis in the future.
References
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823.
- Mueller-Mang, C.; Plank, C.; Ringl, H.; Dirisamer, A.; Herold, C.J. Interstitial Lung Diseases. Med Radiol. 2009, 333–355.
- American Thoracic Society. European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. Am. Thorac. Soc. AJRCCM 2002, 165, 277–304.
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.; Selman, M.; Wells, A.U.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748.
- Antoniou, K.M.; Margaritopoulos, G.A.; Tomassetti, S.; Bonella, F.; Costabel, U.; Poletti, V. Interstitial lung disease. Eur. Respir. Rev. 2014, 23, 40–54.
- Mueller-Mang, C.; Grosse, C.; Schmid, K.; Stiebellehner, L.; Bankier, A.A. What Every Radiologist Should Know about Idiopathic Interstitial Pneumonias. Radiographics 2007, 27, 595–615.
- Kligerman, S.J.; Groshong, S.; Brown, K.K.; Lynch, D.A. Nonspecific Interstitial Pneumonia: Radiologic, Clinical, and Pathologic Considerations. Radiographics 2009, 29, 73–87.
- Siemińska, A.; Kuziemski, K. Respiratory bronchiolitis-interstitial lung disease. Orphanet J. Rare Dis. 2014, 9, 106.
- Cordier, J.-F. Cryptogenic organizing pneumonia. Clin. Chest Med. 2004, 25, 727–738.
- Bruminhent, J.; Yassir, S.; Pippim, J. Acute Interstitial Pneumonia (Hamman-Rich Syndrome) as a Cause of Idiopathic Acute Respiratory Distress Syndrome. Case Rep. Med. 2011, 2011, 1–4.
- A Mikolasch, T.; Garthwaite, H.S.; Porter, J.C. Update in diagnosis and management of interstitial lung disease. Clin. Med. 2017, 17, 146–153.
- Furini, F.; Carnevale, A.; Casoni, G.L.; Guerrini, G.; Cavagna, L.; Govoni, M.; Sciré, C.A. The Role of the Multidisciplinary Evaluation of Interstitial Lung Diseases: Systematic Literature Review of the Current Evidence and Future Perspectives. Front. Med. 2019, 6, 246.
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. [Internet] Frontiers; 2018. Available online: (accessed on 20 January 2021).
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002.
- Shen, H.; Xia, L.; Lu, J. Interleukin-4 in rheumatoid arthritis patients with interstitial lung disease: A pilot study. Indian J. Med. Res. 2013, 138, 919–921.
- Jakubzick, C.; Kunkel, S.L.; Puri, R.K.; Hogaboam, C.M. Therapeutic Targeting of IL-4- and IL-13-Responsive Cells in Pulmonary Fibrosis. Immunol. Res. 2004, 30, 339–350.
- Jakubzick, C.; Choi, E.S.; Carpenter, K.J.; Kunkel, S.L.; Evanoff, H.; Martinez, F.J.; Flaherty, K.R.; Toews, G.B.; Colby, T.V.; Travis, W.D.; et al. Human Pulmonary Fibroblasts Exhibit Altered Interleukin-4 and Interleukin-13 Receptor Subunit Expression in Idiopathic Interstitial Pneumonia. Am. J. Pathol. 2004, 164, 1989–2001.
- Passalacqua, G.; Mincarini, M.; Colombo, D.; Troisi, G.; Ferrari, M.; Bagnasco, D.; Balbi, F.; Riccio, A.M.; Canonica, G.W. IL-13 and idiopathic pulmonary fibrosis: Possible links and new therapeutic strategies. Pulm. Pharmacol. Ther. 2017, 45, 95–100.
- Park, C.S.; Chung, S.W.; Ki, S.Y.; Lim, G.-I.; Uh, S.-T.; Kim, Y.H.; Choi, D.I.; Park, J.S.; Lee, D.W.; Kitaichi, M. Increased Levels of Interleukin-6 Are Associated with Lymphocytosis in Bronchoalveolar Lavage Fluids of Idiopathic Nonspecific Interstitial Pneumonia. Am. J. Respir. Crit. Care Med. 2000, 162, 1162–1168.
- Mozaffarian, A.; Brewer, A.W.; Trueblood, E.S.; Luzina, I.G.; Todd, N.W.; Atamas, S.P.; Arnett, H.A. Mechanisms of Oncostatin M-Induced Pulmonary Inflammation and Fibrosis. J. Immunol. 2008, 181, 7243–7253.
- Ng, B.; Cook, S.A.; Schafer, S. Interleukin-11 signaling underlies fibrosis, parenchymal dysfunction, and chronic inflammation of the airway. Exp. Mol. Med. 2020, 52, 1871–1878.
- Shi, K.; Jiang, J.; Ma, T.; Xie, J.; Duan, L.; Chen, R.; Song, P.; Yu, Z.; Liu, C.; Zhu, Q.; et al. Pathogenesis pathways of idiopathic pulmonary fibrosis in bleomycin-induced lung injury model in mice. Respir. Physiol. Neurobiol. 2014, 190, 113–117.
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811.
- Zhang, Y.; Dees, C.; Beyer, C.; Lin, N.-Y.; Distler, A.; Zerr, P.; Palumbo, K.; Susok, L.; Kreuter, A.; Distler, O.; et al. Inhibition of casein kinase II reduces TGFβ induced fibroblast activation and ameliorates experimental fibrosis. Ann. Rheum. Dis. 2014, 74, 936–943.
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escrivá, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 24.
- Beyer, C.; Distler, J.H.W. Tyrosine kinase signaling in fibrotic disorders: Translation of basic research to human disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 897–904.
- Shaw, R.J.; Benedict, S.H.; Clark, R.A.F.; King, T.E. Pathogenesis of Pulmonary Fibrosis in Interstitial Lung Disease: Alveolar Macrophage PDGF(B) Gene Activation and Up-regulation by Interferon Gamma. Am. Rev. Respir. Dis. 1991, 143, 167–173.
- Homma, S.; Nagaoka, I.; Abe, H.; Takahashi, K.; Seyama, K.; Nukiwa, T.; Kira, S. Localization of platelet-derived growth factor and insulin-like growth factor I in the fibrotic lung. Am. J. Respir. Crit. Care Med. 1995, 152, 2084–2089.
- Sun, Q.; Liu, L.; Mandal, J.; Molino, A.; Stolz, D.; Tamm, M.; Lu, S.; Roth, M. PDGF-BB induces PRMT1 expression through ERK1/2 dependent STAT1 activation and regulates remodeling in primary human lung fibroblasts. Cell Signal 2016, 28, 307–315.
- Chen, Y.; Surinkaew, S.; Naud, P.; Qi, X.-Y.; Gillis, M.-A.; Shi, Y.-F.; Tardif, J.-C.; Dobrev, D.; Nattel, S. JAK-STAT signalling and the atrial fibrillation promoting fibrotic substrate. Cardiovasc. Res. 2017, 113, 310–320.
- Jaskiewicz, K.; Mycroft, K.; Maskey-Warzechowska, M.; Paralusz, K.; Siemiez, N.; Nejman-Gryz, P.; Barnas, M.; Krenke, R.; Gorska, K. Exhaled Biomarkers in Idiopathic Pulmonary Fibrosis—A Six-Month Follow-Up Study in Patients Treated with Pirfenidone. J. Clin. Med. 2020, 9, 2523.
- Simler, N.R.; E Brenchley, P.; Horrocks, A.W.; Greaves, S.M.; Hasleton, P.S.; Egan, J.J. Angiogenic cytokines in patients with idiopathic interstitial pneumonia. Thorax 2004, 59, 581–585.
- Zhou, G.; Xu, Y.; He, B.; Ma, R.; Wang, Y.; Chang, Y.; Xie, Y.; Wu, L.; Huang, J.; Xiao, Z. Ionizing radiation modulates vascular endothelial growth factor expression through STAT3 signaling pathway in rat neonatal primary astrocyte cultures. Brain Behav. 2020, 10, e01529.
- Pedroza, M.; Le, T.T.; Lewis, K.M.; Karmouty-Quintana, H.; To, S.; George, A.T.; Blackburn, M.R.; Tweardy, D.J.; Agarwal, S.K. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016, 30, 129–140.
- Chakraborty, D.; Šumová, B.; Mallano, T.; Chen, C.-W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 2017, 8, 1–16.
- Pechkovsky, D.V.; Prêle, C.M.; Wong, J.; Hogaboam, C.M.; McAnulty, R.; Laurent, G.J.; Zhang, S.S.-M.; Selman, M.; Mutsaers, S.E.; Knight, D.A. STAT3-Mediated Signaling Dysregulates Lung Fibroblast-Myofibroblast Activation and Differentiation in UIP/IPF. Am. J. Pathol. 2012, 180, 1398–1412.
- Chen, Y.-L.; Zhang, X.; Bai, J.; Gai, L.; Ye, X.-L.; Zhang, L.; Xu, Q.; Zhang, Y.-X.; Xu, L.; Li, H.-P.; et al. Sorafenib ameliorates bleomycin-induced pulmonary fibrosis: Potential roles in the inhibition of epithelial–mesenchymal transition and fibroblast activation. Cell Death Dis. 2013, 4, e665.
- Liu, R.-Y.; Zeng, Y.; Lei, Z.; Wang, L.; Yang, H.; Liu, Z.; Zhao, J.; Zhang, H.-T. JAK/STAT3 signaling is required for TGF-β-induced epithelial-mesenchymal transition in lung cancer cells. Int. J. Oncol. 2014, 44, 1643–1651.
- Colomiere, M.; Ward, A.C.; Riley, C.; Trenerry, M.K.; Cameron-Smith, D.; Findlay, J.; Ackland, L.; Ahmed, N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial–mesenchymal transition in ovarian carcinomas. Br. J. Cancer 2008, 100, 134–144.
- Xiao, J.; Gong, Y.; Chen, Y.; Yu, D.; Wang, X.; Zhang, X.; Dou, Y.; Liu, D.; Cheng, G.; Lu, S.; et al. IL-6 promotes epithelial-to-mesenchymal transition of human peritoneal mesothelial cells possibly through the JAK2/STAT3 signaling pathway. Am. J. Physiol. Physiol. 2017, 313, F310–F318.
- Li, M.; Luan, F.; Zhao, Y.; Hao, H.; Zhou, Y.; Han, W.; Fu, X. Epithelial-mesenchymal transition: An emerging target in tissue fibrosis. Exp. Biol. Med. 2016, 241, 1–13.
- Hill, C.; Jones, M.; Davies, D.; Wang, Y. Epithelial-Mesenchymal Transition Contributes to Pulmonary Fibrosis via Aberrant Epithelial/Fibroblastic Cross-Talk. J. Lung Heal. Dis. 2019, 3, 31–35.
- Fintha, A.; Gasparics, Á.; Rosivall, L.; Sebe, A. Therapeutic Targeting of Fibrotic Epithelial-Mesenchymal Transition–An Outstanding Challenge. Front. Pharmacol. 2019, 10, 388.
- Bharadwaj, U.; Kasembeli, M.M.; Robinson, P.; Tweardy, D.J. Targeting Janus Kinases and Signal Transducer and Activator of Transcription 3 to Treat Inflammation, Fibrosis, and Cancer: Rationale, Progress, and Caution. Pharmacol. Rev. 2020, 72, 486–526.
- Waters, D.W.; Blokland, K.; Pathinayake, P.S.; Wei, L.; Schuliga, M.; Jaffar, J.; Westall, G.P.; Hansbro, P.M.; Prele, C.M.; Mutsaers, S.E.; et al. STAT3 Regulates the Onset of Oxidant-induced Senescence in Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 61, 61–73.
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’Cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L1164–L1173.
- Prêle, C.M.; Yao, E.; O’Donoghue, R.J.J.; Mutsaers, S.E.; Knight, D.A. STAT3: A central mediator of pulmonary fibrosis? Proc. Am. Thorac. Soc. 2012, 9, 177–182.
- Moodley, Y.P.; Scaffidi, A.K.; Misso, N.L.; Keerthisingam, C.; McAnulty, R.; Laurent, G.J.; Mutsaers, S.E.; Thompson, P.J.; Knight, D.A. Fibroblasts Isolated from Normal Lungs and Those with Idiopathic Pulmonary Fibrosis Differ in Interleukin-6/gp130-Mediated Cell Signaling and Proliferation. Am. J. Pathol. 2003, 163, 345–354.
- O’Dwyer, D.N.; Ashley, S.L.; Moore, B.B. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L590–L601.
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L56–L69.
- Vasarmidi, E.; Sarantoulaki, S.; Trachalaki, A.; Margaritopoulos, G.; Bibaki, E.; Spandidos, D.; Tzanakis, N.; Antoniou, K. Investigation of key autophagy-and mitophagy-related proteins and gene expression in BALF cells from patients with IPF and RA-ILD. Mol. Med. Rep. 2018, 18, 3891–3897.
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488.
- You, L.; Wang, Z.; Liangkun, Y.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Zhanggui, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739.
- Satarker, S.; Tom, A.A.; Shaji, R.A.; Alosious, A.; Luvis, M.; Nampoothiri, M. JAK-STAT Pathway Inhibition and their Implications in COVID-19 Therapy. Postgrad. Med. 2020, 1–19.
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537.
- Li, X.; Ma, X. Acute Respiratory Failure in COVID-19: Is it “Typical” ARDS? Crit Care [Internet]. 2020. Available online: (accessed on 27 May 2021).
- Atabati, E.; Dehghani-Samani, A.; Mortazavimoghaddam, S.G. Association of COVID-19 and other viral infections with interstitial lung diseases, pulmonary fibrosis, and pulmonary hypertension: A narrative review. Can. J. Respir. Ther. 2020, 56, 70–78.
- Myall, K.J.; Mukherjee, B.; Castanheira, A.M.; Lam, J.L.; Benedetti, G.; Mak, S.M.; Preston, R.; Thillai, M.; Dewar, A.; Molyneaux, P.L.; et al. Persistent Post-COVID-19 Interstitial Lung Disease. An Observational Study of Corticosteroid Treatment. Ann. Am. Thorac. Soc. 2021, 18, 799–806.
- Gagiannis, D.; Steinestel, J.; Hackenbroch, C.; Schreiner, B.; Hannemann, M.; Bloch, W.; Umathum, V.G.; Gebauer, N.; Rother, C.; Stahl, M.; et al. Clinical, Serological, and Histopathological Similarities Between Severe COVID-19 and Acute Exacerbation of Connective Tissue Disease-Associated Interstitial Lung Disease (CTD-ILD). Front. Immunol. 2020, 11, 587517.
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm. Med. 2020, 2020, 1–10.
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815.
- Zhang, C.; Wu, Z.; Li, J.; Tan, K.; Yang, W.; Zhao, H.; Wang, G. Discharge may not be the end of treatment: Pay attention to pulmonary fibrosis caused by severe COVID-19. J. Med. Virol. 2021, 93, 1378–1386.
- Yanhong, R.; Shiyao, W.; Min, L.; Youmin, G.; Huaping, D. When COVID-19 encounters interstitial lung disease: Challenges and management. Chin. J. Tuberc. Respir. Dis. Chin. Med. J. 2020, 43, E039.
- Hui, D.S.; Joynt, G.; Wong, K.T.; Gomersall, C.; Li, T.S.; Antonio, G.; Ko, F.W.S.; Chan, M.C.; Chan, D.P.; Tong, M.W.; et al. Impact of severe acute respiratory syndrome (SARS) on pulmonary function, functional capacity and quality of life in a cohort of survivors. Thorax 2005, 60, 401–409.
- Antonio, G.E.; Wong, K.T.; Hui, D.; Wu, A.; Lee, N.; Yuen, E.H.Y.; Leung, C.B.; Rainer, T.; Cameron, P.; Chung, S.S.C.; et al. Thin-Section CT in Patients with Severe Acute Respiratory Syndrome Following Hospital Discharge: Preliminary Experience. Radiology 2003, 228, 810–815.
- Ong, K.-C.; Ng, A.W.-K.; Lee, L.S.-U.; Kaw, G.; Kwek, S.-K.; Leow, M.K.-S.; Earnest, A. 1-Year Pulmonary Function and Health Status in Survivors of Severe Acute Respiratory Syndrome. Chest 2005, 128, 1393–1400.
- Das, K.M.; Lee, E.Y.; Singh, R.; Enani, M.A.; Al Dossari, K.; Van Gorkom, K.; Larsson, S.G.; Langer, R.D. Follow-up chest radiographic findings in patients with MERS-CoV after recovery. Indian J. Radiol. Imaging 2017, 27, 342–349.
- Luo, J.; Lu, S.; Yu, M.; Zhu, L.; Zhu, C.; Li, C.; Fang, J.; Zhu, X.; Wang, X. The potential involvement of JAK-STAT signaling pathway in the COVID-19 infection assisted by ACE2. Gene 2021, 768, 145325.
- Meletiadis, J.; Tsiodras, S.; Tsirigotis, P. Interleukin-6 Blocking vs. JAK-STAT Inhibition for Prevention of Lung Injury in Patients with COVID-19. Infect. Dis. Ther. 2020, 9, 707–713.
- Cao, Y.; Wei, J.; Zou, L.; Jiang, T.; Wang, G.; Chen, L.; Huang, L.; Meng, F.; Huang, L.; Wang, N.; et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): A multicenter, single-blind, randomized controlled trial. J. Allergy Clin. Immunol. 2020, 146, 137–146.
- Cantini, F.; Niccoli, L.; Matarrese, D.; Nicastri, E.; Stobbione, P.; Goletti, D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J. Infect. 2020, 81, 318–356.
- Zhang, X.; Zhang, Y.; Qiao, W.; Zhang, J.; Qi, Z. Baricitinib, a drug with potential effect to prevent SARS-COV-2 from entering target cells and control cytokine storm induced by COVID-19. Int. Immunopharmacol. 2020, 86, 106749.
- Seif, F.; Aazami, H.; Khoshmirsafa, M.; Kamali, M.; Mohsenzadegan, M.; Pornour, M.; Mansouri, D. JAK Inhibition as a New Treatment Strategy for Patients with COVID-19. Int. Arch. Allergy Immunol. 2020, 181, 467–475.
- Luo, W.; Li, Y.-X.; Jiang, L.-J.; Chen, Q.; Wang, T.; Ye, D.-W. Targeting JAK-STAT Signaling to Control Cytokine Release Syndrome in COVID-19. Trends Pharmacol. Sci. 2020, 41, 531–543.
- Pearce, L.; Davidson, S.M.; Yellon, D.M. The cytokine storm of COVID-19: A spotlight on prevention and protection. Expert Opin. Ther. Targets 2020, 24, 723–730.
- Wang, J.; Wang, B.J.; Yang, J.C.; Wang, M.Y.; Chen, C.; Luo, G.X.; He, W.F. Research advances in the mechanism of pulmonary fibrosis induced by coronavirus disease 2019 and the corresponding therapeutic measures. Zhonghua Shao Shang Za Zhi 2020, 36, 691–697.

