 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Theofilos Kolettis | + 1573 word(s) | 1573 | 2021-05-20 11:40:46 | | | |
| 2 | Lindsay Dong | Meta information modification | 1573 | 2021-06-25 08:00:31 | | |
Video Upload Options
Myocardial infarction often leads to progressive structural and electrophysiologic remodeling of the left ventricle. Histological and functional studies have demonstrated extensive alterations of sympathetic nerve endings at the peri-infarct area and flow-innervation mismatches that create a highly arrhythmogenic milieu.
1. Introduction
Myocardial infarction (MI) often leads to substantial loss of contractile tissue, despite prompt revascularization in the acute phase. Due to the high incidence of coronary artery disease, progressive left ventricular (LV) dilatation and dysfunction post-MI constitutes the most frequent cause of heart failure, which is associated with high morbidity and mortality. Recent evidence demonstrates that heart failure post-MI remains a major health-related problem worldwide, diagnosed in 20–30% of patients one year after the acute event, with the rates steadily rising thereafter [1]. In addition to disabling symptoms and frequent hospitalizations, heart failure is also associated with high incidence of ventricular tachyarrhythmias (VTs), often heralding sudden cardiac death (SCD). The most effective antiarrhythmic strategy is hitherto offered by implantable cardioverter-defibrillators (ICDs), which promptly terminate VTs and prolong survival in selected post-MI populations [2]. Combined with cardiac resynchronization, device-therapy can also ameliorate LV asynchrony and, thereby, improve overall LV performance [3]. However, the value of ICD implantation is hampered by major limitations in current risk stratification algorithms that often fail to identify patients at risk for SCD [4]. As a result, there is an utter need for better understanding of the mechanisms underlying arrhythmogenesis in the post-MI setting, with a view towards expanding the present pharmacologic and non-pharmacologic armamentarium (Figure 1).
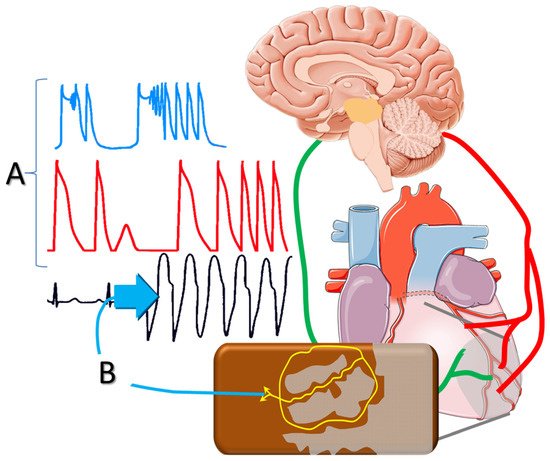
Figure 1. Sympathetic activation post-infarction triggers delayed afterdepolarizations and polymorphic ventricular tachycardia; and (A) facilitates reentrant mechanisms via (B) altering the conduction properties of the peri-infarct tissue.
2. Myocardial Salvage
The extent of myocardial necrosis after acute coronary occlusion and the resultant LV dilatation and dysfunction are the most important predictors of long-term outcome. In this regard, the widespread use of acute reperfusion strategies in recent years has had major impact on the incidence of SCD post-MI [5]. Thrombolysis, and mainly, percutaneous coronary interventions decrease infarct size and transmurality, thereby, ameliorating the substrate for chronic-phase VTs. Therefore, acute MI patients, presenting early in tertiary hospitals, seem to have excellent prognosis during long-term follow-up [6]. However, a substantial proportion of patients with acute MI present too late for myocardial tissue salvage by revascularization; this subgroup, currently estimated at the range of 20%, is characterized by high short- and long-term complication rates [7].
3. Ventricular Remodeling Post-MI
Myocardial necrosis alters loading conditions and triggers a cascade of events, frequently referred to as LV remodeling [8]. Commencing during the early hours after acute coronary occlusion, myocardial loss increases local wall stress in the infarcted area, causing expansion of the border zone [9]. This course is closely intertwined with infarct healing, a dynamic response that initiates a reparative process, leading to collagen scar formation [10]. Structural remodeling is histologically characterized by fibrosis, which is evident not only in the tissue within and adjacent to the infarct, but also in remote myocardial areas.
4. Increased Sympathetic Drive
Chronically elevated sympathetic drive affects a number of ionic currents (referred to as ionic remodeling), resulting in global alteration of LV electrophysiologic properties [11]. For instance, norepinephrine reduces the inward rectifier potassium current, increases resting membrane potential and enhances abnormal automaticity, an action mediated via β-adrenergic receptors. In the presence of a substrate that is invariably located in the peri-infarct tissue, extrasystolic activity may facilitate the onset of monomorphic VTs [12]. Re-entrant mechanisms are also facilitated by chronic β-receptor stimulation via topographic and functional alterations of connexins [13]. Moreover, norepinephrine shortens the duration of the ventricular action potential and elicits delayed afterdepolarizations at high heart rates, thereby, triggering polymorphic VTs that can degenerate into ventricular fibrillation. Last, sympathetic activation affects the restitution features of the ventricular myocardium and alters its refractory period. The latter effects differ, depending on the baseline regional electrophysiologic properties, with refractory period shortening in normal myocardium, as opposed to prolongation in the border zone, surrounding the infarcted tissue. As a result, sympathetic activation can markedly enhance repolarization dispersion in the peri-infarct zone and sets the stage for reentrant mechanisms [14].
5. Antiarrhythmic Effects of β-Blockade
β-blockers exert anti-ischemic actions by reducing myocardial metabolic demand and by prolonging diastolic perfusion time secondary to their actions on the sinus node. Moreover, β-blockade obviates action potential changes and increases the threshold for ventricular fibrillation, counterbalancing the pro-fibrillatory effects of myocardial ischemia or sympathetic activation [15]. Based on these anti-ischemic and anti-arrhythmic properties, β-blockade has been consistently shown to lower SCD rates in post-MI patients.
6. Local Versus Circulating Epinephrine
Sympathetic responses entail two major components, namely increased circulating catecholamines from the adrenal medulla, as well as local norepinephrine release from sympathetic nerve endings, elicited by central activation. The pathophysiologic and clinical significance of such distinction has been largely overlooked, a notion likely attributed to the well-studied downstream-effects of β-blockade. Nevertheless, several pieces of recent data highlight the implications of such disparate electrophysiologic effects of sympathetic activation [16]. For example, (left, right, or bilateral) stellate ganglia stimulation produced distinct patterns of repolarization sequence in the normal porcine heart, assessed by analysis of epicardial and endocardial electrograms [17]. These recordings revealed marked dispersion of repolarization, which was absent when the effects of circulating norepinephrine were examined. Therefore, current evidence suggests that the dispersion of repolarization induced by local sympathetic activation in the peri-infarct area is highly arrhythmogenic, contrasting the more uniform alterations, induced by circulating catecholamines.
7. Flow–Innervation Mismatch in Healed MI
In the process of infarct healing, the area of abnormal myocardial blood flow in the peri-infarct tissue is often surrounded by larger areas, characterized by regional impairment of neuronal catecholamine uptake [18]. Such impairment consists of areas with diverse nerve density and increased neuronal excitability that predisposes to arrhythmogenesis [19].
Interestingly, denervated myocardial regions display excessive responses to catecholamines (Figure 2), contributing to inhomogenous electrophysiologic milieu that favors the formation of reentrant circuits [20]. This apparently paradoxical effect may be the result of enhanced norepinephrine production and overspill in sympathetic nerve endings [21]. The downstream effects are potentiated by abnormal myocardial perfusion in the peri-infarct area that prevents effective blockade of β-adrenergic receptors.
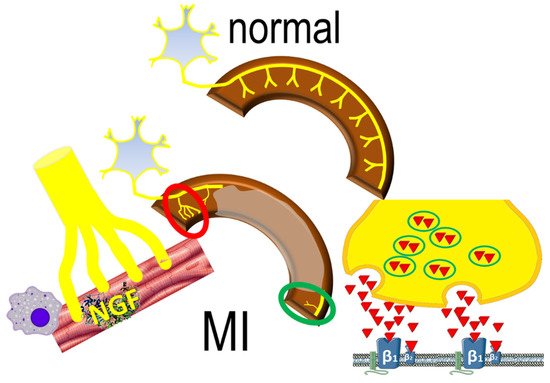
Figure 2. The normal sympathetic nerve distribution (upper panel) is replaced by sympathetic nerve remodeling post-infarct (lower panel), characterized by areas of increased (red circle) sympathetic nerve density, mediated by inflammatory responses; decreased (green circle) nerve density also occurs, characterized by norepinephrine overspill, activating β1 and β2 adrenergic receptors.
8. The Role of Inflammation
Sympathetic nerve remodeling is a complex pathophysiologic process, of which the inflammatory response is thought to be an integral element. Signals from necrotic cells induce the expression of pro-inflammatory cytokines and chemokines that attract neutrophils and monocytes [22]. Activated macrophages play a key-role in sympathetic nerve growth post-MI, exerted mainly via the secretion of nerve growth factor. Inflammatory and autonomic responses may be further interrelated by afferent nerve activation via several cytokines, resulting in efferent modulation of healing responses [23]; the exact structural and biochemical processes are currently under investigation.
9. Central Actions of β-Blockade
The presence of β-adrenergic receptors in the central nervous system has been known for decades. They are located mainly in the hippocampus, the cerebellum, in thalamic nuclei and basal ganglia, as well as in the midbrain and cerebral cortex; low levels of β-receptors are also found in the white matter and hypothalamus [24]. Although the pathophysiologic role of central adrenergic mechanisms in hypertension are well-established [25], their importance in ventricular arrhythmogenesis in the presence of infarcted tissue remains unclear.
10. Autonomic Modulation in Chronic Heart Failure
Based on the detrimental effects of central sympathetic activation, the centrally acting agent moxonidine was evaluated in a multicenter trial [26]. Such incompletely understood drug-effects have shifted research interest towards non-pharmacologic autonomic modulation, with various exercise training regimens presenting as a safe and widely applicable approach [27]. Moreover, in addition to novel pharmaceutical approaches, there is growing interest in interventional procedures, aiming at modulating sympatho-vagal balance [28].
11. Non-Pharmacologic Strategies
A number of non-pharmacologic autonomic interventions are currently under intense investigation in the management of post-MI VTs [29]. Of these, transcutaneous vagal stimulation, stellate ganglion modulation and renal sympathetic denervation seem to have an advantage (Figure 3), as briefly discussed below.
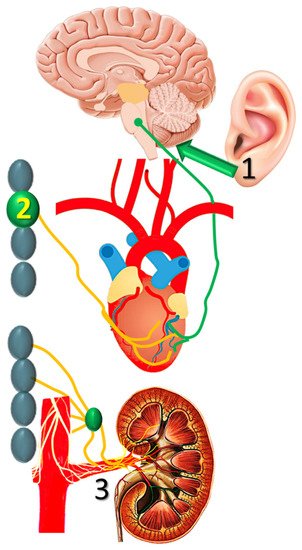
Figure 3. Transcutaneous vagal stimulation, (1) stellate ganglion modulation, and (2) renal sympathetic denervation are (3) currently investigated in nonpharmacologic autonomic modulation post-infarction.
12. Pharmacologic Approaches
In addition to β-blockade, three further treatments have shown efficacy in reducing the incidence of SCD, namely aldosterone blockade, scubitril/valsartan and sodium-glucose co-transporter (type 2) inhibitors. In these paradigms, the issue of ameliorating central sympathetic activation has been raised, strongly reinforcing previous considerations.
13. Summary
Prompt revascularization of the infarct-related coronary artery decreases the necrotic area. Unfortunately, treatment delays are frequent, particularly in elderly patients or diabetics; hence, post-MI heart failure remains a common clinical entity, characterized by evolving structural and electrophysiologic milieu. Despite β-blockade, the incidence of VTs and SCD are high, as current risk-stratification algorithms cannot accurately identify patients that benefit from ICDs.
Several pieces of evidence point towards a crucial role of sympathetic activation on arrhythmogenesis that accompanies progressive post-MI LV remodeling. Examining the complex pathophysiology, a broad spectrum of pre-clinical and clinical studies is currently under way, addressing various aspects of the underlying mechanisms. Non-pharmacologic therapies are intensely investigated, introducing neuromodulatory approaches for post-MI heart failure. Beyond β-blockade, direct pharmacologic approaches were initially disfavored after the disappointing results of moxonidine, but the robust data of SCD reduction by aldosterone antagonists, by sacubitril/valsartan or, more recently, by the sodium-glucose cotransporter type 2 inhibitors have broadened the therapeutic horizon. The pathophysiologic role of the autonomic nervous system is an intriguing topic, with promising clinical applications in the chronic phase of MI.
References
- Jenca, D.; Melenovsky, V.; Stehlik, J.; Stanek, V.; Kettner, J.; Kautzner, J.; Adamkova, V.; Wohlfahrt, P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2021, 8, 222–237.
- Waks, J.W.; Buxton, A.E. Risk stratification for sudden cardiac death after myocardial infarction. Annu. Rev. Med. 2018, 69, 147–164.
- Turitto, G.; El-Sherif, N. Cardiac resynchronization therapy: A review of proarrhythmic and antiarrhythmic mechanisms. Pacing Clin. Electrophysiol. 2007, 30, 115–122.
- Gatzoulis, K.A.; Sideris, A.; Kanoupakis, E.; Sideris, S.; Nikolaou, N.; Antoniou, C.K.; Kolettis, T.M. Arrhythmic risk stratification in heart failure: Time for the next step? Ann. Noninvasive Electrocardiol. 2017, 22, e12430.
- Paranskaya, L.; Akin, I.; Chatterjee, T.; Ritz, A.; Paranski, P.; Rehders, T.; Ince, H.; Schneider, H.; Nienaber, C.A.; Bansch, D. Ventricular tachycardia and sudden death after primary PCI-reperfusion therapy: Impact on primary prevention of sudden cardiac death. Herzschrittmacherther. Elektrophysiol. 2011, 22, 243–248.
- Tran, D.T.; Barake, W.; Galbraith, D.; Norris, C.; Knudtson, M.L.; Kaul, P.; McAlister, F.A.; Sandhu, R.K. Total and cause-specific mortality after percutaneous coronary intervention: Observations from the Alberta provincial project for outcome assessment in Coronary Heart Disease Registry. CJC Open 2019, 1, 182–189.
- McNair, P.W.; Bilchick, K.C.; Keeley, E.C. Very late presentation in ST elevation myocardial infarction: Predictors and long-term mortality. Int. J. Cardiol. Heart Vasc. 2019, 22, 156–159.
- Sutton, M.G.; Sharpe, N. Left ventricular remodeling after myocardial infarction: Pathophysiology and therapy. Circulation 2000, 101, 2981–2988.
- Agathopoulos, S.; Kolettis, T.M. Novel strategies for cardiac repair post-myocardial infarction. Curr. Pharm. Des. 2014, 20, 1925–1929.
- Ertl, G.; Frantz, S. Healing after myocardial infarction. Cardiovasc. Res. 2005, 66, 22–32.
- Dorian, P. Antiarrhythmic action of beta-blockers: Potential mechanisms. J. Cardiovasc. Pharmacol. Ther. 2005, 10 (Suppl. 1), S15–S22.
- Kolettis, T.M. Autonomic function and ventricular tachyarrhythmias during acute myocardial infarction. World J. Exp. Med. 2018, 8, 8–11.
- Campbell, A.S.; Johnstone, S.R.; Baillie, G.S.; Smith, G. β-Adrenergic modulation of myocardial conduction velocity: Connexins vs. sodium current. J. Mol. Cell Cardiol. 2014, 77, 147–154.
- Opthof, T.; Coronel, R.; Vermeulen, J.T.; Verberne, H.J.; van Capelle, F.J.; Janse, M.J. Dispersion of refractoriness in normal and ischaemic canine ventricle: Effects of sympathetic stimulation. Cardiovasc. Res. 1993, 27, 1954–1960.
- Chin, S.H.; Allen, E.; Brack, K.E.; Ng, G.A. Effects of sympatho-vagal interaction on ventricular electrophysiology and their modulation during beta-blockade. J. Mol. Cell Cardiol. 2020, 139, 201–212.
- Fukuda, K.; Kanazawa, H.; Aizawa, Y.; Ardell, J.L.; Shivkumar, K. Cardiac innervation and sudden cardiac death. Circ. Res. 2015, 116, 2005–2019.
- Yagishita, D.; Chui, R.W.; Yamakawa, K.; Rajendran, P.S.; Ajijola, O.A.; Nakamura, K.; So, E.L.; Mahajan, A.; Shivkumar, K.; Vaseghi, M. Sympathetic nerve stimulation, not circulating norepinephrine, modulates T-peak to T-end interval by increasing global dispersion of repolarization. Circ. Arrhythm. Electrophysiol. 2015, 8, 174–185.
- Simoes, M.V.; Barthel, P.; Matsunari, I.; Nekolla, S.G.; Schomig, A.; Schwaiger, M.; Schmidt, G.; Bengel, F.M. Presence of sympathetically denervated but viable myocardium and its electrophysiologic correlates after early revascularised, acute myocardial infarction. Eur. Heart J. 2004, 25, 551–557.
- Li, W.; Knowlton, D.; Van Winkle, D.M.; Habecker, B.A. Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2229–H2236.
- Inoue, H.; Zipes, D.P. Results of sympathetic denervation in the canine heart: Supersensitivity that may be arrhythmogenic. Circulation 1987, 75, 877–887.
- Lorentz, C.U.; Woodward, W.R.; Tharp, K.; Habecker, B.A. Altered norepinephrine content and ventricular function in p75NTR-/- mice after myocardial infarction. Auton. Neurosci. 2011, 164, 13–19.
- Prabhu, S.D.; Frangogiannis, N.G. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ. Res. 2016, 119, 91–112.
- Yin, J.; Wang, Y.; Hu, H.; Li, X.; Xue, M.; Cheng, W.; Wang, Y.; Li, X.; Yang, N.; Shi, Y.; et al. P2X7 receptor inhibition attenuated sympathetic nerve sprouting after myocardial infarction via the NLRP3/IL-1beta pathway. J. Cell Mol. Med. 2017, 21, 2695–2710.
- Reznikoff, G.A.; Manaker, S.; Rhodes, C.H.; Winokur, A.; Rainbow, T.C. Localization and quantification of beta-adrenergic receptors in human brain. Neurology 1986, 36, 1067–1073.
- Koepke, J.P.; DiBona, G.F. Central adrenergic receptor control of renal function in conscious hypertensive rats. Hypertension 1986, 8, 133–141.
- Cohn, J.N.; Pfeffer, M.A.; Rouleau, J.; Sharpe, N.; Swedberg, K.; Straub, M.; Wiltse, C.; Wright, T.J.; Investigators, M. Adverse mortality effect of central sympathetic inhibition with sustained-release moxonidine in patients with heart failure (MOXCON). Eur. J. Heart Fail. 2003, 5, 659–667.
- Hsu, C.Y.; Hsieh, P.L.; Hsiao, S.F.; Chien, M.Y. Effects of exercise training on autonomic function in chronic heart failure: Systematic review. Biomed. Res. Int. 2015, 2015, 591708.
- Franciosi, S.; Perry, F.K.G.; Roston, T.M.; Armstrong, K.R.; Claydon, V.E.; Sanatani, S. The role of the autonomic nervous system in arrhythmias and sudden cardiac death. Auton. Neurosci. 2017, 205, 1–11.
- Lai, Y.; Yu, L.; Jiang, H. Autonomic neuromodulation for preventing and treating ventricular arrhythmias. Front. Physiol. 2019, 10, 200.

