 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yoichiro Otaki | + 1995 word(s) | 1995 | 2021-06-08 09:02:32 | | | |
| 2 | Camila Xu | + 439 word(s) | 2434 | 2021-06-22 11:04:34 | | | | |
| 3 | Camila Xu | + 439 word(s) | 2434 | 2021-06-22 11:05:32 | | |
Video Upload Options
Ubiquitylation is a post-translational modification that covalently conjugates the ubiquitin molecule through the C-terminus to a lysine residue on a substrate protein. Ubiquitylation results in the turnover of the ubiquitylated substrate protein by either the proteasome or lysosome, a change in subcellular localization of the substrate protein, or alteration of substrate protein function . Ubiquitylation is mediated by three enzymes and scaffolding proteins: E1, E2, and E3.
1. Introduction
Despite advances in medicine, cardiovascular disease remains a significant public health problem associated with high mortality [1][2]. Heart failure (HF) is a major cause of cardiovascular deaths. Maladaptive cardiac remodeling caused by hypertension, ischemic heart disease, and other cardiac diseases is accompanied by complex mechanisms that lead to the development of HF [3][4]. Further studies are needed to prevent maladaptive cardiac remodeling and subsequent heart failure.
The modification of eukaryotic proteins with ubiquitin, named ubiquitylation, controls their lifetimes, abundance, localization, interactions, and activities, thereby regulating protein function at all levels. Thus, ubiquitylation plays a pivotal role in a wide range of cellular processes, such as signal transduction, transcriptional regulation, and maintenance of homeostasis. Failing hearts from patients with dilated cardiomyopathy and those with ischemic heart disease show hyper-ubiquitylation compared to donor hearts [5]. The overall observed change in the ubiquitylation cascade in failing hearts is considered an adaptive response to an increased protein burden derived from increased protein synthesis that accompanies the hypertrophic response or an excess of damaged or modified proteins to be targeted for proteasomal degradation.
In the 2000s, many studies focused on the cardiac ubiquitin E3 ligases to clarify the role of ubiquitylation in the development of cardiac diseases, such as the carboxyl terminus of Hsp70 interacting protein (CHIP), atrogen-1, muscle ring finger (MuRF) family, mouse double mutant 2 homolog (MDM2), cellular inhibitor of apoptosis, casitas b-lineage lymphoma, and E6-associated protein (E6AP) [6][7][8][9][10][11][12][13]. Cardiac ubiquitin E3 ligase plays several roles in protein turnover, energy metabolism, receptor internalization, hypertrophic response, apoptosis, and tolerance to ischemia/reperfusion (I/R) in cardiomyocytes [14][15]. Although elevated expression levels of E6AP were observed in mice after pressure overload [12], the functional role of E6AP has never been examined. Thus, our knowledge of the molecular mechanism of HECT-type E3 ligase in the development of cardiac disease is still lacking.
HECT-type E3 ligase is reported to be involved in a wide range of human diseases and health including neurodegenerative diseases, neurological syndromes, and cancers [16][17][18]. HECT-type E3 ligases are highly conserved between cells and tissues; as a result, it is tempting to speculate that they also contribute to human cardiac health and disease. There are only a few review articles summarizing recent advancements regarding HECT-type E3 ligase in the field of cardiac disease [19]. This study focused on cardiac remodeling and described the role of HECT-type E3 ligases in the development of cardiac disease.
2. Ubiquitylation
Ubiquitylation results in the turnover of the ubiquitylated substrate protein by either the proteasome or lysosome, a change in subcellular localization of the substrate protein, or alteration of substrate protein function [20]. Ubiquitylation is mediated by three enzymes and scaffolding proteins: E1, E2, and E3. There are only few E1s and several E2s; however, E3 ligases constitute a large class of proteins with the human genome encoding more than 600 putative E3 ligases and E3 ligase complexes [21][22][23][24]. E3 ligases are also modulators of the rate-limiting step in this enzymatic cascade, participating in substrate protein recognition and catalytic transfer of ubiquitin.
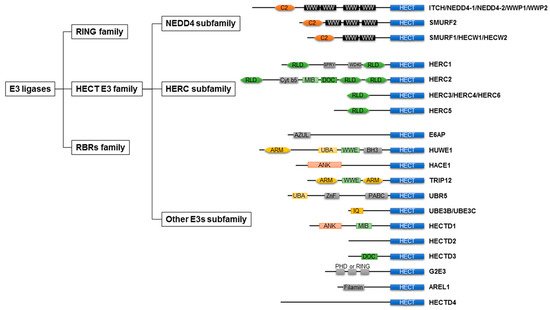
As shown in Figure 1, E3 ligases are classified into three groups: really interesting new genes (RING), homologous to E6AP C-terminus (HECT), and RING-between-RINGs (RBRs). The domain architecture and mechanism of ubiquitylation depend on the class of E3 ligases [25].
Substrate proteins are modified by a single ubiquitin moiety on one or multiple sites, giving rise to mono- and multi-mono-ubiquitylated proteins, respectively. In addition, a wide variety of polyubiquitin chains can be formed on substrate proteins, in which the ubiquitin moieties can be linked through either one of the seven internal lysine residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63) in ubiquitin or through its N-terminal amino group.
Polyubiquitylation through the Lsy48-linked ubiquitin chain is generally used for the ubiquitin–proteasomal degradation pathway. Substrate proteins that receive Lys48-linked polyubiquitin chains migrate to and are degraded by the 26S proteasome. The ubiquitin–proteasome system is a protein quality and quantity control system that mediates approximately 80–90% of intracellular protein degradation under optimal nutritional conditions [26][27][28][29][30]. Furthermore, mono-ubiquitylation of lysine residues or polyubiquitylation through Lys63-linked ubiquitin chains are used for nonproteolytic pathways such as DNA repair, relocalization, modifying activity (signal transcriptional activity), or endocytosis [31][32][33].
RING E3 ligases catalyze the direct transfer of ubiquitin from the E2 conjugating enzyme to the substrate, suggesting that the linkage type of the ubiquitin chain is determined by the E2 conjugating enzyme. In contrast to RING-type E3 ligases, HECT-type E3 ligases include an active-site cysteine in the HECT domain, which forms an intermediate thioester bond with ubiquitin before it is conjugated to the substrate protein [34][35]. HECT-type E3 ligase has enzymatic activity and directly catalyzes the covalent attachment of ubiquitin to substrate proteins; therefore, it could determine the linkage type of ubiquitin chain preferred [36].
E6AP transcribed from the ubiquitin–protein ligase There are 28 types of HECT-type E3 ligases in humans [37], which are commonly grouped into three groups based on the presence of distinct amino acid sequence motifs or domains within the N-terminal: NEDD4 subfamily, HERC subfamily, and other HECT-type E3 ligases [38] (Figure 1). The N-lobe represents the E2 binding domain, whereas the C-lobe contains an active site cysteine to receive ubiquitin. In the HECT family, 16–92% amino acid identity was found for this domain [39].
The NEDD4 subfamily member includes nine types of HECT-type E3 ligases and accounts for approximately 30% of HECT-type E3 ligases [18]. The N-terminal C2 domain is defined as a Ca+ phospholipid binder [40]. The WW domains are responsible for recognizing substrates and have also been found to form intramolecular interactions with the HECT domain of the E3 ligases [41][42]. Some NEDD4 subfamily members are often expressed as alternative splice isoforms [38].
The HERC subfamily is characterized by a HECT domain and one or more regulators of chromosome condensation-like domains (RLDs), an effector protein domain that was first identified as a regulator of chromosome condensation 1 [43]. In humans, the HERC subfamily comprises six members, which can be further organized into two large and four small HERCs. Large HERCs (HERC1 and 2) have two or three RLDs; however, small HERCs (HERCs 3, 4, 5, and 6) have one RLD. RLD has dual functions: one side of the domain acts as a guanine nucleotide exchange factor for the small GTPase Ran, whereas the opposite side interacts with chromatin through histones H2A and H2B [44][45].
Each member of another HECT E3 ligase lacks WW or RLD domains and has a distinct variety of N-terminal domains. There are several N-terminal domains of other HECT E3 ligases, such as WWE and armadillo repeats (HUWE1 and TRIP12), , ankyrin repeats ( HACE1 and HECTD1), and IQ motifs (UBE3B and UBE3C)
3. Importance of Ubiquitylation in Cardiac Disease
Accumulating evidence indicates that ubiquitylation is involved in developing cardiac diseases [13][46][47][48][49][50]. Cardiac proteins are in a dynamic state of continual degradation and resynthesis and are thought to replace all in 30 days under normal circumstances. An experimental study demonstrated that the balance of protein turnover could lead to protein accumulation and aggravation during cardiac remodeling [51]. The discovery that cardiac ubiquitin E3 ligases, such as muscle-specific ubiquitin ligase atrogin-1 and MuRF family, yields cardiac growth, and remodeling through sarcomeric protein turnover, indicated that the ubiquitylation cascade is fundamental to the maintenance of normal cardiac function through protein quality control [13][52][53].
As previously mentioned, ubiquitylation is involved in most aspects of eukaryotic cell biology, such as intracellular signaling, transcriptional control, and regulation of cell death. Research regarding the role of cardiac ubiquitin E3 ligases has developed from protein turnover to cellular processes such as signal transduction, transcriptional regulation, maintenance of homeostasis, mitochondrial dynamics, receptor turnover, and energy metabolism [13][15]. In the following paragraph, we will limit ourselves to a discussion of HECT-type E3 ligases that have been associated with cardiac diseases.
4. Cardiac Fibrosis and HECT-Type E3 Ligase
Pathological cardiac fibrosis is a process characterized by excessive deposition of extracellular matrix (ECM), leading to the development of cardiac dysfunction, arrhythmia, and HF [54][55][56]. Several pathophysiological conditions induce cardiac fibrosis, such as pressure overload, volume overload, myocardial infarction, dilated and hypertrophied cardiomyopathy, various toxic insults, metabolic disturbances, and aging [57][58][59][60].
Cardiac fibroblasts are key effector cells in cardiac fibrosis and are responsible for ECM homeostasis in the heart [54]. After cardiac fibroblasts are activated by regulators of tissue fibrosis, such as angiotensin II, connective tissue growth factor, bone morphogenetic protein (BMP), Wnt ligands, cytokines, and TGF- TGF-β1 contributes to cardiac fibrosis development through SMAD-dependent and SMAD-independent pathways. TGF-β1 generally exerts its biological effects by activating downstream mediators, including SMAD2 and SMAD3, while negatively regulated by SMAD7 expression [61][62][63].
Although the pathophysiological conditions leading to cardiac fibrosis are different from those of cardiac diseases, it is valuable to explore the common mechanisms involved in cardiac fibrosis. This study shows the current understanding of the role of HECT-type E3 ligases in cardiac fibrosis. An overview of HECT-type E3 ligases in cardiac fibrosis is summarized in Table 1.
Table 1. HECT-type E3 ligase and cardiac remodeling.
| HECT-Type E3 Ligase | Substrate/Target | Main Findings | Reference |
|---|---|---|---|
| Cardiac hypertrophy | |||
| ITCH | Dishevelled | Cardiac-specific ITCH transgenic mice inhibited maladaptive hypertrophy via Wnt/β catenin signal inhibition. | [64] |
| NEDD4-2 | ENaC in kidney | Cardiac hypertrophy was observed in NEDD4-2 null mice on chronic high-salt diet. | [65][66] |
| Circular RNA WWP1 | ANF and miR-23a | Circular RNA WWP1 was dysregulated in the heart treated with isoproterenol. | [67] |
| WWP2 | PARP1 | WWP2 conditional knockout mice (MycCre+;WWP2Fl/Fl) exacerbated isoproterenol-induced cardiac hypertrophy. | [68] |
| E6AP | Increased myocardium E6AP expression after pressure overload. | [12] | |
| HUWE1 | c-myc | HUWE1 conditional knockout mice spontaneously developed cardiac hypertrophy. | [69] |
| HACE1 | Unknown | HACE1 conditional knockout mice spontaneously developed cardiac hypertrophy. | [70] |
| HECTD3 | SUMO2/STAT1 | AAV9-medited overexpression of HECTD3 inhibited pathological hypertrophy in mice. | [71] |
| Cardiac fibrosis | |||
| WWP2 | SMAD2 | WWP2mut/mut mice attenuated cardiac fibrosis after angiotensin II infusion and myocardial infarction. | [72] |
| SMURF1 | SMURF1 was involved in BMP-2 antagonization for TGF-β1 signal. SMURF1 was a target of miR-10b-5p, which inhibits cardiac fibroblast activation. |
[73][74] | |
| SMURF2 | SMAD7 | Mediator of TGF-β signal. SMURF2 mediated SMAD7 degradation was inhibited by SMAD3 inhibitor. |
[75][76] |
| HFpEF | |||
| WWP1 | Not described | Cardiac-specific overexpression of WWP1 developed cardiac hypertrophy with diastolic dysfunction. | [77] |
Chen et al. identified the WWP2 N-terminal isoform as a positive regulator of the pro-fibrotic gene network associated with cardiac fibrosis using systems genetics in human and murine dilated cardiomyopathy and repaired tetralogy of Fallot. The left ventricular single-cell RNA sequence indicated that WWP2 is mainly expressed in fibroblasts, immune cells, and endothelial cells. WWP2mut/mut mice lacking the N-terminal isoform and full-length WWP2 attenuated cardiac fibrosis and preserved cardiac function after angiotensin II infusion or myocardial infarction. These findings provide new understanding into the role of HECT-type E3 ligases independently of the HECT domain.
SMAD ubiquitin regulatory factor (SMURF) was initially identified as a regulator of SMAD1 stability [78]. SMURFs have been implicated in determining the competence of cells in response to the TGF-β/BMP signaling pathway [79]. SMURF is a multifunctional protein that is involved in cell cycle progression, cell proliferation, differentiation, DNA damage response, and maintenance of genomic stability. SMURF1 plays an important role in heart development, including outflow tract septation and cell-type specification, by controlling cilium-associated BMP signaling [80][81].
BMP-2, as a novel fibrosis-antagonizing cytokine, have a potential beneficial effect in attenuating pressure overload-induced cardiac fibrosis. Wang S et al. demonstrated that SMURF1 interacted with SMAD6 and that this SMURF1/SMAD6 complex was involved in BMP2 antagonization of TGF-β1 mediated protein kinase C-δ and SMAD3 signaling in cardiomyocytes [73]. This finding suggests that SMURF1 may contribute to cardiac fibrosis development.
Endothelial colony-forming cells have been reported to reduce cardiac fibrosis in myocardial infarction due to their proliferation and secretion of exosomes, which transfer microRNAs. Cardiac fibroblast activation is ameliorated by exosomes from endothelial colony-forming cells treated with normoxia compared to those treated with hypoxia. Liu et al. found that miR-10b-5p was enriched in exosomes from normoxia and targeted SMURF1 and histone deacetylase 4 using next-generation RNA sequencing. Thus, inhibition of mRNA expression of SMURF1 by miR-10b-5p was suggested to participate in the antifibrotic effects of exosomes derived from endothelial colony-forming cells treated with normoxia [74].
SMURF2 consists of a C2 domain, three WW domains, and an HECT domain. SMURF2 targets SMADs, heat shock proteins 27, and p53 [82]. SMURF2 was reported to be downregulated in DCM [83]. SMURF functions as a mediator of TGF-β signaling via interaction with SMAD7 containing PY motif during cardiac fibrosis [75].
The protein expression level of SMURF2 in the mouse heart increased, while that of SMAD7 decreased after angiotensin II administration. However, this effect was reversed by the SMAD3 inhibitor, suggesting that the SMAD3 inhibitor protected cardiac SMAD7 from SMURF2-mediated ubiquitin–proteasome degradation. Since SMAD7 functions as an inhibitor of both TGF-β/SMAD and NF-κB signaling, an increase in cardiac SMAD7 could be another mechanism through which SMAD3 inhibitor blocked SMAD3-mediated cardiac fibrosis and NF-κB-driven cardiac inflammation [76]. This finding suggests that SMRUF2 may contribute to the development of cardiac fibrosis.
References
- Chen, J.; Normand, S.L.; Wang, Y.; Krumholz, H.M. National and regional trends in heart failure hospitalization and mortality rates for Medicare beneficiaries, 1998–2008. JAMA 2011, 306, 1669–1678.
- Lloyd-Jones, D.M.; Larson, M.G.; Leip, E.P.; Beiser, A.; D’Agostino, R.B.; Kannel, W.B.; Murabito, J.M.; Vasan, R.S.; Benjamin, E.J.; Levy, D. Lifetime risk for developing congestive heart failure: The Framingham Heart Study. Circulation 2002, 106, 3068–3072.
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407.
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 2016, 97, 245–262.
- Weekes, J.; Morrison, K.; Mullen, A.; Wait, R.; Barton, P.; Dunn, M.J. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 2003, 3, 208–216.
- Naito, A.T.; Okada, S.; Minamino, T.; Iwanaga, K.; Liu, M.L.; Sumida, T.; Nomura, S.; Sahara, N.; Mizoroki, T.; Takashima, A.; et al. Promotion of CHIP-mediated p53 degradation protects the heart from ischemic injury. Circ. Res. 2010, 106, 1692–1702.
- Schisler, J.C.; Rubel, C.E.; Zhang, C.; Lockyer, P.; Cyr, D.M.; Patterson, C. CHIP protects against cardiac pressure overload through regulation of AMPK. J. Clin. Investig. 2013, 123, 3588–3599.
- Le, N.T.; Takei, Y.; Shishido, T.; Woo, C.H.; Chang, E.; Heo, K.S.; Lee, H.; Lu, Y.; Morrell, C.; Oikawa, M.; et al. p90RSK targets the ERK5-CHIP ubiquitin E3 ligase activity in diabetic hearts and promotes cardiac apoptosis and dysfunction. Circ. Res. 2012, 110, 536–550.
- Arya, R.; Kedar, V.; Hwang, J.R.; McDonough, H.; Li, H.H.; Taylor, J.; Patterson, C. Muscle ring finger protein-1 inhibits PKC activation and prevents cardiomyocyte hypertrophy. J. Cell Biol. 2004, 167, 1147–1159.
- Willis, M.S.; Schisler, J.C.; Li, L.; Rodriguez, J.E.; Hilliard, E.G.; Charles, P.C.; Patterson, C. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ. Res. 2009, 105, 80–88.
- Toth, A.; Nickson, P.; Qin, L.L.; Erhardt, P. Differential regulation of cardiomyocyte survival and hypertrophy by MDM2, an E3 ubiquitin ligase. J. Biol. Chem. 2006, 281, 3679–3689.
- Balasubramanian, S.; Mani, S.; Shiraishi, H.; Johnston, R.K.; Yamane, K.; Willey, C.D.; Cooper, G., 4th; Tuxworth, W.J.; Kuppuswamy, D. Enhanced ubiquitination of cytoskeletal proteins in pressure overloaded myocardium is accompanied by changes in specific E3 ligases. J. Mol. Cell Cardiol. 2006, 41, 669–679.
- Willis, M.S.; Bevilacqua, A.; Pulinilkunnil, T.; Kienesberger, P.; Tannu, M.; Patterson, C. The role of ubiquitin ligases in cardiac disease. J. Mol. Cell Cardiol. 2014, 71, 43–53.
- Zolk, O.; Schenke, C.; Sarikas, A. The ubiquitin-proteasome system: Focus on the heart. Cardiovasc. Res. 2006, 70, 410–421.
- Parry, T.L.; Willis, M.S. Cardiac ubiquitin ligases: Their role in cardiac metabolism, autophagy, cardioprotection and therapeutic potential. Biochim. Biophys. Acta 2016, 1862, 2259–2269.
- Wang, Y.; Argiles-Castillo, D.; Kane, E.I.; Zhou, A.; Spratt, D.E. HECT E3 ubiquitin ligases-emerging insights into their biological roles and disease relevance. J. Cell Sci. 2020, 133.
- Mao, X.; Sethi, G.; Zhang, Z.; Wang, Q. The Emerging Roles of the HERC Ubiquitin Ligases in Cancer. Curr. Pharm. Des. 2018, 24, 1676–1681.
- Scheffner, M.; Kumar, S. Mammalian HECT ubiquitin-protein ligases: Biological and pathophysiological aspects. Biochim. Biophys. Acta 2014, 1843, 61–74.
- Zhang, Y.; Qian, H.; Wu, B.; You, S.; Wu, S.; Lu, S.; Wang, P.; Cao, L.; Zhang, N.; Sun, Y. E3 Ubiquitin ligase NEDD4 familyregulatory network in cardiovascular disease. Int. J. Biol. Sci. 2020, 16, 2727–2740.
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253.
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537.
- Varshavsky, A. The ubiquitin system, an immense realm. Annu. Rev. Biochem. 2012, 81, 167–176.
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180.
- Hutchins, A.P.; Liu, S.; Diez, D.; Miranda-Saavedra, D. The repertoires of ubiquitinating and deubiquitinating enzymes in eukaryotic genomes. Mol. Biol. Evol. 2013, 30, 1172–1187.
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157.
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403.
- Lyon, R.C.; Lange, S.; Sheikh, F. Breaking down protein degradation mechanisms in cardiac muscle. Trends Mol. Med. 2013, 19, 239–249.
- Portbury, A.L.; Willis, M.S.; Patterson, C. Tearin’ up my heart: Proteolysis in the cardiac sarcomere. J. Biol. Chem. 2011, 286, 9929–9934.
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899.
- Schubert, U.; Antón, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774.
- Peralta, D.A.; Araya, A.; Busi, M.V.; Gomez-Casati, D.F. The E3 ubiquitin-ligase SEVEN IN ABSENTIA like 7 mono-ubiquitinates glyceraldehyde-3-phosphate dehydrogenase 1 isoform in vitro and is required for its nuclear localization in Arabidopsis thaliana. Int. J. Biochem. Cell Biol. 2016, 70, 48–56.
- Xie, F.; Zhang, Z.; van Dam, H.; Zhang, L.; Zhou, F. Regulation of TGF-β Superfamily Signaling by SMAD Mono-Ubiquitination. Cells 2014, 3, 981–993.
- Welchman, R.L.; Gordon, C.; Mayer, R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 2005, 6, 599–609.
- Huibregtse, J.M.; Scheffner, M.; Beaudenon, S.; Howley, P.M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA 1995, 92, 2563–2567.
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 1995, 373, 81–83.
- Kim, H.C.; Huibregtse, J.M. Polyubiquitination by HECT E3s and the determinants of chain type specificity. Mol. Cell Biol. 2009, 29, 3307–3318.
- Scheffner, M.; Staub, O. HECT E3s and human disease. BMC Biochem. 2007, 8 (Suppl. 1), S6.
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409.
- Zhang, W.; Wu, K.P.; Sartori, M.A.; Kamadurai, H.B.; Ordureau, A.; Jiang, C.; Mercredi, P.Y.; Murchie, R.; Hu, J.; Persaud, A.; et al. System-Wide Modulation of HECT E3 Ligases with Selective Ubiquitin Variant Probes. Mol. Cell 2016, 62, 121–136.
- Rizo, J.; Sudhof, T.C. C2-domains, structure and function of a universal Ca2+-binding domain. J. Biol. Chem. 1998, 273, 15879–15882.
- Ingham, R.J.; Colwill, K.; Howard, C.; Dettwiler, S.; Lim, C.S.; Yu, J.; Hersi, K.; Raaijmakers, J.; Gish, G.; Mbamalu, G.; et al. WW domains provide a platform for the assembly of multiprotein networks. Mol. Cell Biol. 2005, 25, 7092–7106.
- Riling, C.; Kamadurai, H.; Kumar, S.; O’Leary, C.E.; Wu, K.P.; Manion, E.E.; Ying, M.; Schulman, B.A.; Oliver, P.M. Itch WW Domains Inhibit Its E3 Ubiquitin Ligase Activity by Blocking E2-E3 Ligase Trans-thiolation. J. Biol. Chem. 2015, 290, 23875–23887.
- Bischoff, F.R.; Ponstingl, H. Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature 1991, 354, 80–82.
- Zhang, C.; Clarke, P.R. Chromatin-independent nuclear envelope assembly induced by Ran GTPase in Xenopus egg extracts. Science 2000, 288, 1429–1432.
- Nemergut, M.E.; Mizzen, C.A.; Stukenberg, T.; Allis, C.D.; Macara, I.G. Chromatin docking and exchange activity enhancement of RCC1 by histones H2A and H2B. Science 2001, 292, 1540–1543.
- Willis, M.S.; Patterson, C. Proteotoxicity and cardiac dysfunction--Alzheimer’s disease of the heart? N. Engl. J. Med. 2013, 368, 455–464.
- Patterson, C.; Ike, C.; Willis, P.W., 4th; Stouffer, G.A.; Willis, M.S. The bitter end: The ubiquitin-proteasome system and cardiac dysfunction. Circulation 2007, 115, 1456–1463.
- Willis, M.S.; Patterson, C. Into the heart: The emerging role of the ubiquitin-proteasome system. J. Mol. Cell Cardiol. 2006, 41, 567–579.
- Powell, S.R. The ubiquitin-proteasome system in cardiac physiology and pathology. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1–H19.
- Brown, D.I.; Parry, T.L.; Willis, M.S. Ubiquitin Ligases and Posttranslational Regulation of Energy in the Heart: The Hand that Feeds. Compr. Physiol. 2017, 7, 841–862.
- Tsukamoto, O.; Minamino, T.; Kitakaze, M. Functional alterations of cardiac proteasomes under physiological and pathological conditions. Cardiovasc. Res. 2010, 85, 339–346.
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708.
- Li, H.H.; Kedar, V.; Zhang, C.; McDonough, H.; Arya, R.; Wang, D.Z.; Patterson, C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J. Clin. Investig. 2004, 114, 1058–1071.
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040.
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 2007, 117, 568–575.
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2020.
- Ashrafian, H.; McKenna, W.J.; Watkins, H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ. Res. 2011, 109, 86–96.
- Bernaba, B.N.; Chan, J.B.; Lai, C.K.; Fishbein, M.C. Pathology of late-onset anthracycline cardiomyopathy. Cardiovasc. Pathol. 2010, 19, 308–311.
- Asbun, J.; Villarreal, F.J. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J. Am. Coll Cardiol. 2006, 47, 693–700.
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574.
- Watanabe, K.; Narumi, T.; Watanabe, T.; Otaki, Y.; Takahashi, T.; Aono, T.; Goto, J.; Toshima, T.; Sugai, T.; Wanezaki, M.; et al. The association between microRNA-21 and hypertension-induced cardiac remodeling. PLoS ONE 2020, 15, e0226053.
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-beta Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461.
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into TGF-beta/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018, 292, 76–83.
- Goto, J.; Otaki, Y.; Watanabe, T.; Kobayashi, Y.; Aono, T.; Watanabe, K.; Wanezaki, M.; Kutsuzawa, D.; Kato, S.; Tamura, H.; et al. HECT (Homologous to the E6-AP Carboxyl Terminus)-Type Ubiquitin E3 Ligase ITCH Attenuates Cardiac Hypertrophy by Suppressing the Wnt/beta-Catenin Signaling Pathway. Hypertension 2020, 76, 1868–1878.
- Galiana-Simal, A.; Olivares-Alvaro, E.; Klett-Mingo, M.; Ruiz-Roso, M.B.; Ballesteros, S.; de Las Heras, N.; Fuller, P.J.; Lahera, V.; Martin-Fernandez, B. Proanthocyanidins block aldosterone-dependent up-regulation of cardiac gamma ENaC and Nedd4-2 inactivation via SGK1. J. Nutr. Biochem. 2016, 37, 13–19.
- Shi, P.P.; Cao, X.R.; Sweezer, E.M.; Kinney, T.S.; Williams, N.R.; Husted, R.F.; Nair, R.; Weiss, R.M.; Williamson, R.A.; Sigmund, C.D.; et al. Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am. J. Physiol. Renal. Physiol. 2008, 295, F462–F470.
- Yang, M.H.; Wang, H.; Han, S.N.; Jia, X.; Zhang, S.; Dai, F.F.; Zhou, M.J.; Yin, Z.; Wang, T.Q.; Zang, M.X.; et al. Circular RNA expression in isoproterenol hydrochloride-induced cardiac hypertrophy. Aging 2020, 12, 2530–2544.
- Zhang, N.; Zhang, Y.; Qian, H.; Wu, S.; Cao, L.; Sun, Y. Selective targeting of ubiquitination and degradation of PARP1 by E3 ubiquitin ligase WWP2 regulates isoproterenol-induced cardiac remodeling. Cell Death Differ. 2020, 27, 2605–2619.
- Dadson, K.; Hauck, L.; Hao, Z.; Grothe, D.; Rao, V.; Mak, T.W.; Billia, F. The E3 ligase Mule protects the heart against oxidative stress and mitochondrial dysfunction through Myc-dependent inactivation of Pgc-1alpha and Pink1. Sci. Rep. 2017, 7, 41490.
- Zhang, L.; Chen, X.; Sharma, P.; Moon, M.; Sheftel, A.D.; Dawood, F.; Nghiem, M.P.; Wu, J.; Li, R.K.; Gramolini, A.O.; et al. HACE1-dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat. Commun. 2014, 5, 3430.
- Rangrez, A.Y.; Borlepawar, A.; Schmiedel, N.; Deshpande, A.; Remes, A.; Kumari, M.; Bernt, A.; Christen, L.; Helbig, A.; Jungmann, A.; et al. The E3 ubiquitin ligase HectD3 attenuates cardiac hypertrophy and inflammation in mice. Commun. Biol. 2020, 3, 562.
- Chen, H.; Moreno-Moral, A.; Pesce, F.; Devapragash, N.; Mancini, M.; Heng, E.L.; Rotival, M.; Srivastava, P.K.; Harmston, N.; Shkura, K.; et al. WWP2 regulates pathological cardiac fibrosis by modulating SMAD2 signaling. Nat. Commun. 2019, 10, 3616.
- Wang, S.; Sun, A.; Li, L.; Zhao, G.; Jia, J.; Wang, K.; Ge, J.; Zou, Y. Up-regulation of BMP-2 antagonizes TGF-β1/ROCK-enhanced cardiac fibrotic signalling through activation of Smurf1/Smad6 complex. J. Cell Mol. Med. 2012, 16, 2301–2310.
- Liu, W.; Zhang, H.; Mai, J.; Chen, Z.; Huang, T.; Wang, S.; Chen, Y.; Wang, J. Distinct Anti-Fibrotic Effects of Exosomes Derived from Endothelial Colony-Forming Cells Cultured Under Normoxia and Hypoxia. Med. Sci. Monit. 2018, 24, 6187–6199.
- Cunnington, R.H.; Nazari, M.; Dixon, I.M. c-Ski, Smurf2, and Arkadia as regulators of TGF-beta signaling: New targets for managing myofibroblast function and cardiac fibrosis. Can. J. Physiol. Pharmacol. 2009, 87, 764–772.
- Meng, J.; Qin, Y.; Chen, J.; Wei, L.; Huang, X.R.; Yu, X.; Lan, H.Y. Treatment of Hypertensive Heart Disease by Targeting Smad3 Signaling in Mice. Mol. Ther. Methods Clin. Dev. 2020, 18, 791–802.
- Matesic, L.E.; Freeburg, L.A.; Snyder, L.B.; Duncan, L.A.; Moore, A.; Perreault, P.E.; Zellars, K.N.; Goldsmith, E.C.; Spinale, F.G. The ubiquitin ligase WWP1 contributes to shifts in matrix proteolytic profiles and a myocardial aging phenotype with diastolic heart. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H765–H774.
- Zhu, H.; Kavsak, P.; Abdollah, S.; Wrana, J.L.; Thomsen, G.H. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 1999, 400, 687–693.
- Lin, X.; Liang, M.; Feng, X.H. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J. Biol. Chem. 2000, 275, 36818–36822.
- Duenas, A.; Exposito, A.; Munoz, M.D.M.; de Manuel, M.J.; Camara-Morales, A.; Serrano-Osorio, F.; Garcia-Padilla, C.; Hernandez-Torres, F.; Dominguez, J.N.; Aranega, A.; et al. MiR-195 enhances cardiomyogenic differentiation of the proepicardium/septum transversum by Smurf1 and Foxp1 modulation. Sci. Rep. 2020, 10, 9334.
- Koefoed, K.; Skat-Rørdam, J.; Andersen, P.; Warzecha, C.B.; Pye, M.; Andersen, T.A.; Ajbro, K.D.; Bendsen, E.; Narimatsu, M.; Vilhardt, F.; et al. The E3 ubiquitin ligase SMURF1 regulates cell-fate specification and outflow tract septation during mammalian heart development. Sci. Rep. 2018, 8, 9542.
- David, D.; Nair, S.A.; Pillai, M.R. Smurf E3 ubiquitin ligases at the cross roads of oncogenesis and tumor suppression. Biochim. Biophys. Acta 2013, 1835, 119–128.
- Molina-Navarro, M.M.; Trivino, J.C.; Martinez-Dolz, L.; Lago, F.; Gonzalez-Juanatey, J.R.; Portoles, M.; Rivera, M. Functional networks of nucleocytoplasmic transport-related genes differentiate ischemic and dilated cardiomyopathies. A new therapeutic opportunity. PLoS ONE 2014, 9, e104709.

