Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sangmyung Rhee | + 3587 word(s) | 3587 | 2021-06-17 05:03:04 | | | |
| 2 | Bruce Ren | -21 word(s) | 3566 | 2021-06-22 09:35:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rhee, S. Microtubule Acetylation Controls Breast Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/11114 (accessed on 04 July 2026).
Rhee S. Microtubule Acetylation Controls Breast Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/11114. Accessed July 04, 2026.
Rhee, Sangmyung. "Microtubule Acetylation Controls Breast Cancer" Encyclopedia, https://encyclopedia.pub/entry/11114 (accessed July 04, 2026).
Rhee, S. (2021, June 22). Microtubule Acetylation Controls Breast Cancer. In Encyclopedia. https://encyclopedia.pub/entry/11114
Rhee, Sangmyung. "Microtubule Acetylation Controls Breast Cancer." Encyclopedia. Web. 22 June, 2021.
Copy Citation
During aggressive cancer progression, cancer cells adapt to unique microenvironments by withstanding various cellular stresses, including endoplasmic reticulum (ER) stress. However, the mechanism whereby cancer cells overcome the ER stress to survive remains to be elucidated.
breast cancer
microtubule acetylation
extracellular matrix
ER stress
unfolded protein response
1. Introduction
The tumor microenvironment is composed of cells, including cancer cells, immune cells, and fibroblasts, and acellular components surrounding the cancer cells, such as the extracellular matrix (ECM) and factors secreted by the cells such as growth factors, metabolites, etc. This unique microenvironment promotes cancer cell growth and metastasis. The deposition of ECM molecules in various solid cancers, including breast cancer, causes biomechanical and biochemical changes in the cancer microenvironment during cancer progression. Studies have indicated that the tumor mass is up to ten times harder or denser than normal tissue [1][2]. In general, with the progression of cancers, the stiffness of the extracellular matrices surrounding cancer cells increases; this causes the cancer cells to become more aggressive, along with massive cytoskeletal rearrangement and alterations in oncogenic signals. For example, cancer cells cultured on a stiff matrix showed increased cytoskeletal tension, resulting in enhanced cell–ECM adhesion, disruption of cell–cell junctions, and increased cell proliferation, compared to those of cells cultured on a soft matrix [3]. Enhanced cell–matrix adhesion initiates the recruitment of focal adhesion signaling molecules, such as focal adhesion kinase, proto-oncogene non-receptor tyrosine kinase src (Src), and paxillin, leading to integrin clustering and promoting cancer progression [4][5]. Moreover, an increased collagen matrix density promotes the invasiveness of breast epithelial cells [6]. These aggressive traits of cancer cells are the result of their successful adaptation to various cellular stresses in response to biomechanical and biochemical changes in the cancer microenvironment.
The accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER) induces ER stress, which triggers the unfolded protein response (UPR) to restore protein homeostasis [7]. In ER stress conditions, binding immunoglobin protein (BiP/GRP78) interacts with unfolded proteins, thus activating UPR signaling [8]. In cancer, ER stress promotes cancer development by triggering the UPR signaling molecules such as protein kinase RNA-like endoplasmic reticulum kinase (PERK) and activating transcription factor (ATF) 6 [7][9][10], whereas prolonged and excessive ER stress can induce apoptosis through the inositol-requiring enzyme-1α (IRE1α)-mediated signaling pathway [11][12]. Thus, a better understanding of the mechanisms that regulate ER stress in cancer cells will provide important clues for cancer treatment.
Microtubules are major components of the eukaryotic cytoskeleton and are involved in various cellular processes, such as cell division, motility, and cellular transport [13][14]. The cellular functions of microtubules are mostly governed by post-translational modifications. Especially, acetylation at lysine 40 (K40) of α-tubulin in long-lived or stable microtubules is critical for cancer progression. In basal-like and metastatic breast cancers, microtubule acetylation is increased and induces microtentacle formation, cell adhesion, migration, and invasion during vascular traveling [15]. In addition, increased microtubule acetylation by α-tubulin N-acetyltransferase 1 (αTAT1) promotes the invasiveness of colon cancer cells via Wnt/β-catenin signaling [16], and microtubule hyperacetylation through sirtuin 2 (SIRT2) inhibition promotes the proliferation of cancer cells and the growth of tumors [17]. It has been recently reported that the microtubule-disrupting agent N-deacetyl-N-(chromone-2-carbonyl)-thiocolchicine activates several transcription factors, including ATF6, ATF4, ATF3, and CCAAT-enhancer binding protein homologous protein (CHOP) [18]. This implies that microtubules are mechanistically linked to apoptotic cell death via the ER stress pathway. However, the role of αTAT1 in the regulation of ER stress in cancer cells is presently unknown.
2. Increased ECM Stiffness Inhibits ER Stress
To identify differentially expressed genes (DEGs) according to ECM stiffness, we performed RNA-sequencing (RNA-seq) analysis of MDA-MB-231 breast cancer cells cultured on a collagen-coated 0.5 kPa polyacrylamide gel (soft matrix) or a collagen-coated culture dish (stiff matrix). RNA-seq data sets were obtained from independent biological duplicates. To identify congruency between each biological replicate, principal component analysis (PCA) of transcripts using DEseq2 was performed, which indicated that the biological replicates from each experimental group were clustered to identify high reproducibility between the replicates (Figure 1A). Pearson’s correlation analysis of RNA-seq data also showed high correlation with over 99.9% similarity between replicates in the soft and stiff matrix groups . As shown in Figure 1B, a volcano plot was constructed by integrating both the p-value and fold change of each transcript (p-value ≤ 0.05 and absolute log2 (fold change) ≥ 1.5) to indicate the general scattering of the transcripts and to filter the differentially expressed transcripts for different cellular environments. In total, 985 DEGs (RNA-seq FPKM values having log2 (fold change) ≥ 1.5, adjusted p-value < 0.05, average FPKM in each group ≥ 20%) were identified. The results were presented as heatmap expression based on the unsupervised hierarchical clustering of expression ratios for all DEGs (Figure 1C).
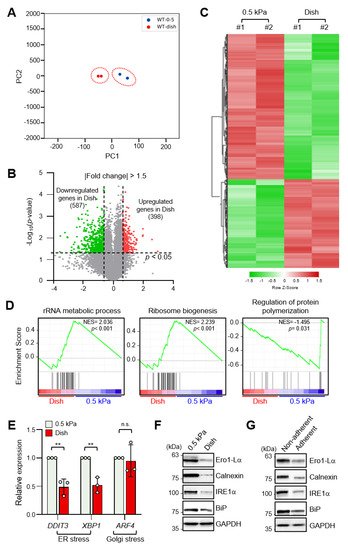
Figure 1. Endoplasmic reticulum (ER) stress is differentially regulated by extracellular matrix (ECM) stiffness. (A) Principal component analysis (PCA) of the RNA-sequencing (RNA-seq) datasets obtained from MDA-MB-231 cells cultured on soft and stiff matrices. (B) Volcano plot of differentially expressed genes (DEGs) in MDA-MB-231 cells according to ECM stiffness based on RNA-seq results. Genes with significantly decreased expression are shown in green; genes with significantly increased expression are shown in red. (C) Heatmap of all DEGs between cells cultured on soft matrix and stiff matrix. (D) Gene set enrichment analysis (GSEA) of ECM stiffness-dependent signaling pathways. (E) Expression of ER stress and Golgi stress markers in MDA-MB-231 cells cultured on 0.5 kPa polyacrylamide gels (PAGs) or culture dishes was quantified by RT-qPCR. Values represent the mean ± SD of three independent experiments and were analyzed using Student’s t-test. ** p < 0.01, N.S. not significant. (F) MDA-MB-231 cells were cultured on 0.5 kPa PAGs or dishes for 48 h. Cell lysates were used for Western blotting analysis of the ER stress markers Ero1-Lα, Calnexin (CANX), IRE1α, and BiP. (G) MDA-MB-231 cells were cultured on non-adherent or adherent plates for 48 h. Cell lysates were used for Western blotting analysis of the ER stress markers Ero1-Lα, Calnexin, IRE1α, and BiP. GAPDH was analyzed as a loading control in all Western blot assays.
Gene set enrichment analysis (GSEA) of all DEGs showed that genes involved in the rRNA metabolic process and the ribosome biogenesis pathway were enriched in cells grown on a stiff matrix, whereas genes involved in the regulation of protein polymerization were enriched in cells cultured on a soft matrix (Figure 1D). This finding suggested that protein synthesis is more robust in cells cultured on a stiff matrix than in cells cultured on a soft matrix, which is in line with previous findings [19].
As it has been reported that ER stress triggers homeostatic regulation of protein synthesis in response to extra- and intracellular stress signals [20][21], we next compared ER stress marker expression in MDA-MB-231 cells according to matrix stiffness. Interestingly, transcript levels of DDIT3 and XBP1s, but not the Golgi stress marker ATF4, were significantly lower in cells grown on a stiff matrix than in cells grown on a soft matrix (Figure 1E). In addition, protein levels of the ER stress markers Ero1-Lα, calnexin (CANX), PERK, IRE1α, and BiP were also significantly lower under the stiff environment (Figure 1F indicating that ER stress is substantially lower in the former. MDA-MB-231 cells cultured in non-adherent dishes also showed increased expression of the ER stress markers (Figure 1G). These results confirmed that external mechanical stress applied to cells affects ER stress.
3. Microtubule Acetylation Is Required for the Regulation of UPR Signaling
As the spreading of MDA-MB-231 cells was significantly increased on a stiff matrix compared to a soft matrix as previously published [22], we examined whether microtubule acetylation is also required for MDA-MB-231 cell spreading on a stiff matrix. The level of microtubule acetylation, but not detyrosination, was increased in cells cultured on a stiff matrix compared with that in cells cultured on a soft matrix (Figure 2A). In addition, microtubule acetylation was downregulated in MDA-MB-231 cells cultured on non-adherent dishes compared to that in cells cultured on adherent dishes (Figure 2B). However, cells incubated with Y-27632 and blebbistatin, a ROCK inhibitor, and myosin II inhibitor to reduce the cellular tension did not show changes in the levels of microtubule acetylation. Collectively, these results indicated that microtubule acetylation is likely induced by cell–substrate adhesion but not by actomyosin contractility during cell spreading.
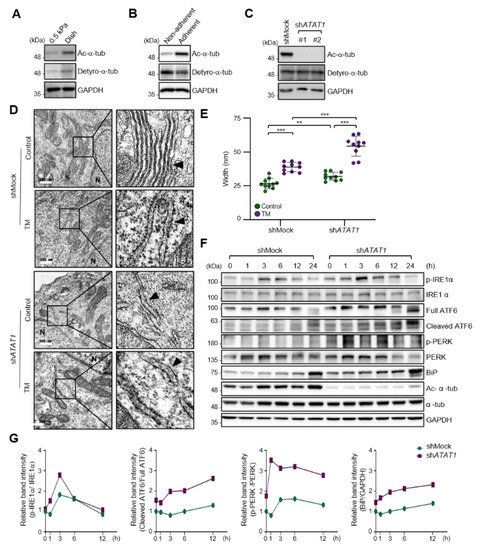
Figure 2. ECM stiffness-dependent microtubule acetylation regulates ER stress response signaling. (A) Western blot analysis of acetylated and detyrosinated α-tubulin levels in MDA-MB-231 cells cultured on 0.5 kPa PAGs or culture dishes for 48 h. (B) Western blot analysis of acetylated and detyrosinated α-tubulin levels in MDA-MB-231 cells cultured on non-adherent or adherent plates for 48 h. (C) Western blot analysis of ATAT1 KD efficiency in shATAT1 #1- and #2-treated MDA-MB-231 cells compared to shMock-treated cells. (D) Transmission electron microscopy of shMock- and shATAT1 #1-treated MDA-MB-231 cells in the presence or absence of 20 ng/mL tunicamycin (TM) for 24 h. The ER is indicated by arrowheads. Scale bar, 500 nm. (E) Morphometric analysis of ER width in shMock- and shATAT1 #1-treated MDA-MB-231 cells (n = 10). Statistical analysis was performed using one-way ANOVA followed by Tukey multiple comparison tests. One-way ANOVA, F3, 36 = 63.35. ** p < 0.01, *** p < 0.001. (F) Western blot analysis of phospho-IRE1α, IRE1α, ATF6, phospho-PERK, PERK, BiP, acetylated α-tubulin, and α-tubulin in shMock- and shATAT1 #1-treated MDA-MB-231 cells treated with 20 ng/mL TM for the indicated times. GAPDH was analyzed as a loading control in all Western blot assays. (G) Quantification of relative expression in UPR and ER stress marker proteins shown in (F). Band intensities of target proteins were quantified by densitometry using a Quantity One® system. Relative expression of phospho-IRE1α, ATF6, and phospho-PERK were normalized by the band intensities of total IRE1a, full ATF6, and total PERK, respectively. The relative expression of Bip was normalized with GAPDH band intensity. The Western blot images are representative images of the results of at least three independent biological replicates.
As cells on stiff and adherent matrices had acetylated microtubules and lower ER stress marker expression, we hypothesized that microtubule acetylation is involved in the downregulation of ER stress markers in cells grown on a stiff matrix. To test this hypothesis, we established ATAT1 knockdown (KD) MDA-MB-231 cells using shRNA, which had a ~90% reduction in microtubule acetylation when grown on a stiff matrix (Figure 2C). The ER of ATAT1 KD cells had an abnormal structure, including features such as the widening and disruption of the alignments of the ER cisternae compared to the control cells; these abnormal structures were similar to those typically observed in tunicamycin-treated cells showing increased ER stress levels (Figure 2D,E). Treatment with tunicamycin further increased the degree of structural abnormalities of the ER in ATAT1 KD cells (Figure 2D,E). UPR signals, including IRE1α phosphorylation, ATF6 cleavage, and the expression of ER stress markers such as BiP, were also significantly increased in ATAT1 KD cells after tunicamycin treatment (Figure 2F,G). These results suggested that microtubule acetylation is involved in the regulation of UPR signaling under ER stress conditions.
4. Regulation of Cancer Pathway-Related Gene Expression by ER Stress in a Stiff Matrix Is Dependent on Microtubule Acetylation
To determine alterations in signaling pathways according to ECM stiffness, we conducted a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of 398 DEGs whose expression levels were increased in cells grown on a stiff matrix when compared to cells grown on a soft matrix, as shown in Figure 1C. “Pathways in cancer” was the most enriched pathway, with 17 genes (Figure 3A). We have previously reported that TGF-β-induced microtubule acetylation in fibroblasts regulates gene expression by promoting the nuclear translocation of yes-associated protein [23]. Therefore, to ascertain whether the 17 genes upregulated in cells grown on a stiff matrix were also transcriptionally regulated by microtubule acetylation, we constructed the ATAT1 knockout (KO) cell line using the CRISPR/Cas9 technique and performed RNA-seq analysis in WT and ATAT1 KO cells. Interestingly, KEGG pathway analysis of 3145 DEGs whose expression levels were lower in ATAT1 KO cells than that in WT cells showed that “Pathways in cancer” was also the most enriched pathway. Moreover, among the 17 genes obtained from Figure 3A, 10 genes (MYC, MITF, MAPK8, CXCR4, RASSF1, ETS1, BCL2, CXCL8, FGF1, and BMP2) were downregulated and three genes (E2F2, CCNE1, and LPAR1) were upregulated in ATAT1 KO cells, while others (CYCS, E2F1, WNT10B, and RAD51) were not significantly changed in their expression between mock and KO samples (Figure 3B). Among them, 7 of 10 downregulated genes were confirmed to be significantly downregulated by quantitative reverse transcription (RT-q) PCR analysis (Figure 3C).
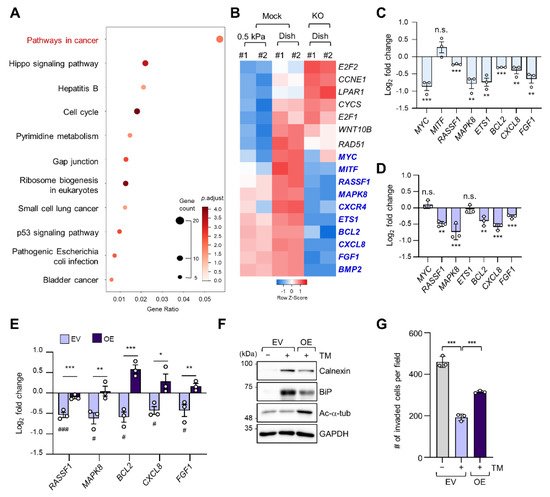
Figure 3. Microtubule acetylation induces cancer-related gene expression through the alleviation of ER stress in breast cancer cells. (A) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of DEGs upregulated in cells grown on a stiff matrix. The x-axis indicates the gene ratio, i.e., the ratio of DEGs in the given gene ontology (GO) term. The y-axis indicates KEGG pathways. Dot size represents the number of genes in each KEGG pathway. (B) Heatmap of 17 genes related to “Pathways in cancer” obtained through KEGG pathway analysis in mock and ATAT1 KO MDA-MB-231 cells. Genes with reduced expression in ATAT1 KO compared to mock-treated cells are indicated by blue letters. (C) mRNA levels of “Pathways in cancer” genes were decreased in ATAT1 KO MDA-MB-231 cells treated with 20 ng/mL tunicamycin for 24 h as indicated by RT-qPCR. Values represent the mean ± SD of three independent experiments. ** p < 0.01, *** p < 0.001, N.S. not significant (Student’s t-test). (D) mRNA levels of seven genes that were downregulated upon tunicamycin treatment shown in (C) in ATAT1 KD compared to shMock-treated MDA-MB-231 cells as determined by RT-qPCR. Values represent the mean ± SD of three independent experiments. ** p < 0.01, *** p < 0.001, N.S. not significant (Student’s t-test). (E) mRNA levels of “Pathway in cancer” genes in control and ATAT1 overexpression lines after tunicamycin treatment as assessed by RT-qPCR. Values represent the mean ± SD of three independent experiments. Statistical significance of the differences in gene expression according to tunicamycin treatment in each of the cell lines transfected with empty vectors (EV) and ATAT1 overexpression (OE) vectors was analyzed by one-way ANOVA followed by Tukey multiple comparison tests (# p < 0.05, ### p < 0.001). One-way ANOVA, F2, 6 = 46.21 (RASSF1), F2, 6 = 12.70 (MAPK8), F2, 6 = 38.36 (BCL2), F2, 6 = 9.008 (CXCL8), and F2, 6 = 10.71 (FGF1). Statistical significance of the differences between EV controls and ATAT1 OE cells was also analyzed using Student’s t-test (* p < 0.05, ** p < 0.01, *** p < 0.001). (F) Western blot analysis of calnexin (CANX), BiP, and acetylated α-tubulin in EV controls or ATAT1 OE cells after treatment with 20 ng/mL tunicamycin for 24 h. GAPDH was used as loading control. (G) Comparison of the number of invading cells via a Transwell invasion assay in cells cultured under the same conditions as in (F). Values represent the mean ± SD of three independent experiments. One-way ANOVA, F2, 6 = 158.4. *** p < 0.001.
As we found that microtubule acetylation is involved in the modulation of ER stress intensity, we investigated whether the expression of the seven cancer-related genes regulated by microtubule acetylation was also affected by ER stress. Five out of seven genes were also downregulated upon tunicamycin treatment (Figure 3D). Interestingly, their expression was restored upon overexpression of ATAT1 (Figure 3E).
To confirm whether the above results were due to reduced ER stress, we analyzed ER stress marker expression and cell invasion of ATAT1-overexpressing cells after tunicamycin treatment. In ATAT1-overexpressing cells, tunicamycin-induced ER stress marker expression was decreased when compared to that in control cells, and cell invasion was increased (Figure 3F,G). These results suggested that microtubule acetylation modulates ER stress in cells grown on a stiff matrix, thereby increasing the expression of genes involved in cancer signaling pathways, promoting breast cancer progression, including invasiveness.
5. Microtubule Acetylation and ER Stress Regulate Focal Adhesion Formation
As microtubule acetylation restored tunicamycin-inhibited MDA-MB-231 cell invasion (Figure 3G), we next evaluated the expression of genes associated with cell migration by Gene Ontology (GO)-term analysis using RNA-seq data from control and ATAT1 KO cells. Figure 4A shows that genes related to focal adhesion and cell adhesion were significantly downregulated in ATAT1 KO cells (Figure 4A). Especially, the expression of VCL, PTK2, PXN, and TLN1, which encode components of focal adhesions, was decreased in ATAT1 KO cells (Figure 4B). ATAT1 KD cells also showed reduced expression levels of focal adhesion proteins, as observed in shMock cells treated with tunicamycin (Figure 4C). In accordance herewith, immunocytochemistry results showed that the number and size of focal adhesions at the pericellular region were reduced in both tunicamycin-treated and ATAT1 KD cells (Figure 4D). Real-time microscopy revealed that the newly formed focal adhesions in response to the FBS stimulation were less dynamic in ATAT1 KD and tunicamycin-treated cells (Figure 4E,F and Figure S7). Taken together, these results indicated that microtubule acetylation controls the expression of focal adhesion proteins and the dynamics of newly formed focal adhesions through the modulation of ER stress, thereby affecting cell migration and invasion.
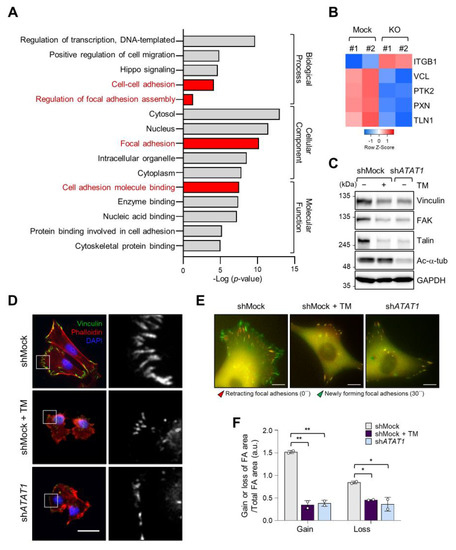
Figure 4. Focal adhesion assembly is regulated by microtubule acetylation and ER stress. (A) Functional annotation of 389 DEGs in ATAT1 KO MDA-MB-231 cells using PANTHER gene ontology (GO). (B) Heatmap showing genes related to focal adhesion assembly based on RNA-seq data from ATAT1 KO cells. (C) Validation of gene expression using Western blotting. shMock-treated cells treated or not with tunicamycin, and shATAT1-treated cells were cultured for 24 h. Cell lysates were used for Western blot analysis of vinculin, FAK, talin, and acetylated α-tubulin, using GAPDH as a loading control. (D) Immunocytochemistry analysis of focal adhesions using antibodies against vinculin and F-actin in shMock-treated and ATAT1 KO cells cultured in the presence of 20 ng/mL tunicamycin for 24 h. Scale bar, 30 μm. (E) Paxillin-GFP-expressing shMock- and shATAT1 #1-treated MDA-MB-231 cells were starved for 16 h in serum-free RPMI1640 medium and then stimulated with 10% FBS with or without 20 ng/mL tunicamycin. Merged paxillin–GFP images in shMock- and shATAT1 #1-treated cells at 0 and 30 min. Red represents retracting focal adhesions and green represents newly forming focal adhesions. Scale bar, 10 μm. (F) Ratios of gain and loss of focal adhesions to total focal adhesion area. Values represent the mean ± SD of two independent experiments. Statistical significances were analyzed using one-way ANOVA followed by Tukey multiple comparison tests. One-way ANOVA (Loss; F2, 3 = 15.98. * p < 0.05, Gain; F2, 3 = 174.1, ** p < 0.01).
6. Expression of ATAT1 and ER Stress Markers Is Negatively Correlated in Breast Cancer Patients
To investigate the correlation between breast cancer progression and microtubule acetylation, we analyzed the level of microtubule acetylation in cancer and normal tissues by performing immunohistochemistry using a commercially available human breast carcinoma tissue microarray. Assuming that a sample with a staining intensity score of 2 or 3 has high amounts of acetylated microtubules, 23/40 (57%) of breast carcinoma specimens were scored as 2 and 3, while 2/10 (20%) had scores of 2 and 3 in the normal and cancer adjacent normal tissues. Consequently, the median of acetylated microtubule intensity was three-fold higher in cancer than in normal tissues (Figure 5A). We next analyzed the level of ATAT1 transcripts in various breast cancer tissues using the Oncomine v4.5 database (https://www.oncomine.org). The ATAT1 transcript was increased in most cancer tissues when compared to the levels in normal tissues (Figure 5B). To further investigate the relationship between ATAT1 transcript levels and ER stress markers in breast cancer, we analyzed the correlation between ATAT1 and ER stress marker gene expression levels using the bc-GenExMiner v4.6 database (http://bcgenex.ico.unicancer.fr). Pearson’s correlation analysis showed that although the correlation coefficient between ATAT1 and ER stress marker genes (except for HSPA5) was low, they tended to have a negative correlation, whereas those of ER stress marker genes were positively correlated (Figure 5C).
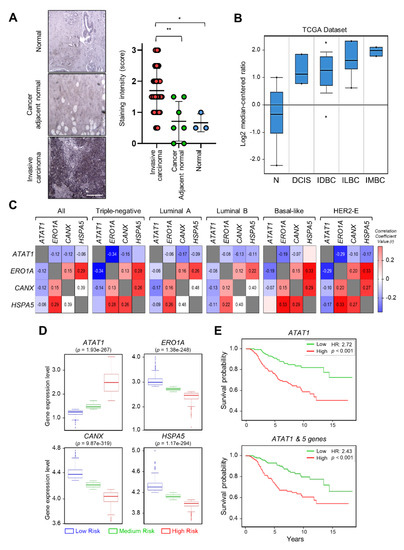
Figure 5. Expression levels of ATAT1 and ER stress marker genes are negatively correlated in breast cancer patients. (A) Representative images of immunohistochemistry of acetylated α-tubulin in normal, cancer-adjacent normal, and invasive carcinoma breast tissues. Scale bar, 200 μm. Lower panels, sample distribution by acetylated α-tubulin staining intensity. Staining intensity was marked (0 = absence, 1 = weak, 2 = moderate, 3 = strong). Values represent the mean ± SD of three independent experiments. Statistical significance was analyzed using one-way ANOVA followed by Tukey’s multiple comparison tests (* p < 0.05, ** p < 0.01). One-way ANOVA (F2, 47 = 8.552, p < 0.001). (B) Analysis of ATAT1 expression levels in normal breast, ductal breast carcinoma in situ, invasive ductal breast carcinoma, invasive lobular breast carcinoma, and invasive mixed breast carcinoma tissues using the Oncomine database. N, normal breast; DCIS, Ductal breast carcinoma in situ; IDBC, invasive ductal breast carcinoma; ILBC, invasive lobular breast carcinoma; IMBC, invasive mixed breast carcinoma. (C) Pearson’s correlations between mRNA levels of ATAT1 and ER stress marker genes in breast cancer patients based on the bc-GenExMiner RNA-seq dataset (n = 4712). Number in the box indicates correlation coefficient value. (D) Expression levels of ATAT1 and ER stress marker genes in low-, medium-, and high-risk groups in 962 breast cancer patients from a TCGA dataset. (E) Kaplan–Meier plots of breast cancer patients based on the expression of ATAT1 and MAPK8, RASSF1, BCL2, CXCL8, and FGF1 in SurvExpress data (n = 295). HR, hazard ratio.
To evaluate the prognostic value of ATAT1 and ER stress marker gene expression in breast cancer patient survival, we utilized the SurvExpress database v2.0 (http://bioinformatica.mty.itesm.mx:8080/Biomatec/SurvivaX.jsp). In high-risk groups, ATAT1 mRNA expression was elevated, whereas the expression of ER stress marker genes such as ERO1A, CANX, and HSPA5, was lower (Figure 5D). In addition, breast cancer patients with high expression of ATAT1 and MAPK8, RASSF1, BCL2, CXCL8, and FGF1, had a poor prognosis (Figure 5E). These results indicated that combinatorial analysis of ER stress marker and ATAT1 transcripts may be useful for the diagnosis of breast cancer progression.
References
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122.
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906.
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254.
- Shi, Q.; Boettiger, D. A novel mode for integrin-mediated signaling: Tethering is required for phosphorylation of FAK Y397. Mol. Biol. Cell 2003, 14, 4306–4315.
- Lawson, C.D.; Burridge, K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 2014, 5, e27958.
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene 2009, 28, 4326–4343.
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2016, 2, 252–262.
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108.
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88.
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597.
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355.
- Yang, W.; Tiffany-Castiglioni, E.; Koh, H.C.; Son, I.H. Paraquat activates the IRE1/ASK1/JNK cascade associated with apoptosis in human neuroblastoma SH-SY5Y cells. Toxicol. Lett. 2009, 191, 203–210.
- Gadde, S.; Heald, R. Mechanisms and molecules of the mitotic spindle. Curr. Biol. 2004, 14, R797–R805.
- Rogers, S.L.; Gelfand, V.I. Membrane trafficking, organelle transport, and the cytoskeleton. Curr. Opin. Cell Biol. 2000, 12, 57–62.
- Boggs, A.E.; Vitolo, M.I.; Whipple, R.A.; Charpentier, M.S.; Goloubeva, O.G.; Ioffe, O.B.; Tuttle, K.C.; Slovic, J.; Lu, Y.; Mills, G.B.; et al. alpha-Tubulin acetylation elevated in metastatic and basal-like breast cancer cells promotes microtentacle formation, adhesion, and invasive migration. Cancer Res. 2015, 75, 203–215.
- Oh, S.; You, E.; Ko, P.; Jeong, J.; Keum, S.; Rhee, S. Genetic disruption of tubulin acetyltransferase, alphaTAT1, inhibits proliferation and invasion of colon cancer cells through decreases in Wnt1/beta-catenin signaling. Biochem. Biophys. Res. Commun. 2017, 482, 8–14.
- Lee, J.K.; Lee, J.; Go, H.; Lee, C.G.; Kim, S.; Kim, H.S.; Cho, H.; Choi, K.S.; Ha, G.H.; Lee, C.W. Oncogenic microtubule hyperacetylation through BEX4-mediated sirtuin 2 inhibition. Cell Death Dis. 2016, 7, e2336.
- Ho, C.T.; Chang, Y.J.; Yang, L.X.; Wei, P.L.; Liu, T.Z.; Liu, J.J. A novel microtubule-disrupting agent induces endoplasmic reticular stress-mediated cell death in human hepatocellular carcinoma cells. PLoS ONE 2015, 10, e0136340.
- Tilghman, R.W.; Blais, E.M.; Cowan, C.R.; Sherman, N.E.; Grigera, P.R.; Jeffery, E.D.; Fox, J.W.; Blackman, B.R.; Tschumperlin, D.J.; Papin, J.A.; et al. Matrix rigidity regulates cancer cell growth by modulating cellular metabolism and protein synthesis. PLoS ONE 2012, 7, e37231.
- Lebeaupin, C.; Vallee, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947.
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011.
- Ko, P.; Kim, D.; You, E.; Jung, J.; Oh, S.; Kim, J.; Lee, K.H.; Rhee, S. Extracellular matrix rigidity-dependent sphingosine-1-phosphate secretion regulates metastatic cancer cell invasion and adhesion. Sci. Rep. 2016, 6, 21564.
- You, E.; Ko, P.; Jeong, J.; Keum, S.; Kim, J.-W.; Seo, Y.-J.; Song, W.K.; Rhee, S. Dynein-mediated nuclear translocation of yes-associated protein through microtubule acetylation controls fibroblast activation. Cell. Mol. Life Sci. 2020, 77, 4143–4161.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
710
Revisions:
2 times
(View History)
Update Date:
22 Jun 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No