 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kazuyuki Nakagome | + 1847 word(s) | 1426 | 2021-06-09 04:15:23 | | | |
| 2 | Vivi Li | Meta information modification | 1847 | 2021-06-21 04:35:57 | | |
Video Upload Options
Eosinophilic pneumonia (EP), including acute EP and chronic EP, is characterized by the massive pulmonary infiltration of eosinophils into the lung. However, the mechanisms underlying the selective accumulation of eosinophils in EP have not yet been fully elucidated. We reported that bronchoalveolar lavage fluid (BALF) from EP patients induced the transmigration of eosinophils across endothelial cells in vitro. The concentrations of eotaxin-2 (CCL24) and monocyte chemotactic protein (MCP)-4 (CCL13), which are CC chemokine receptor (CCR) 3 ligands, were elevated in the BALF of EP patients, and anti-CCR3 monoclonal antibody inhibited the eosinophil transmigration induced by the BALF of EP patients. The concentration of macrophage inflammatory protein 1β (CCL4), a CCR5 ligand that induces eosinophil migration, was increased in the BALF of EP patients. Furthermore, the concentration of interleukin (IL) 5 was increased in the BALF of EP patients, and it has been reported that anti-IL-5 antibody treatment resulted in remission and the reduction of glucocorticoid use in some cases of chronic EP. The concentrations of lipid mediators, such as leukotriene (LT) B4, damage-associated molecular pattern molecules (DAMPs), such as uric acid, or extracellular matrix proteins, such as periostin, were also increased in the BALF of EP patients. These findings suggest that chemokines, such as CCR3/CCR5 ligands, cytokines, such as IL-5, lipid mediators, such as LTB4, DAMPs, and extracellular matrix proteins may play roles in the accumulation or activation of eosinophils in EP.
1. Introduction
2. Mechanisms for the Development of Eosinophilic Airway Inflammation
3. Role of CCR3 Ligands or CCR5 Ligands in the Eosinophil Accumulation in EP
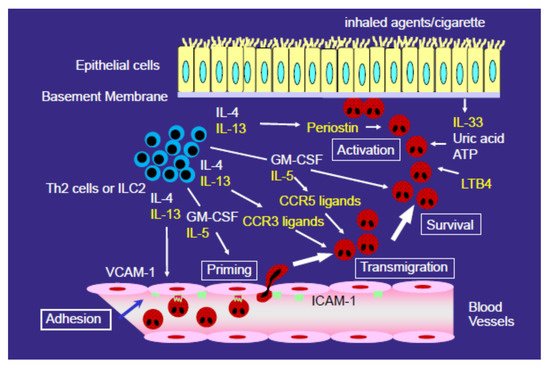
4. Role of Type 2 Cytokines (IL-4 and IL-13) in the Development of EP
References
- Cottin, V.; Cordier, J.F. Eosinophilic pneumonias. Allergy 2005, 60, 841–857.
- Suzuki, Y.; Suda, T. Eosinophilic pneumonia: A review of the previous literature, causes, diagnosis, and management. Allergol. Int. 2019, 68, 413–419.
- Allen, J.N.; Pacht, E.R.; Gadek, J.E.; Davis, W.B. Acute eosinophilic pneumonia as a reversible cause of noninfectious respiratory failure. N. Engl. J. Med. 1989, 321, 569–574.
- Carrington, C.B.; Addington, W.W.; Goff, A.M.; Madoff, I.M.; Marks, A.; Schwaber, J.R.; Gaensler, E.A. Chronic eosinophilic pneumonia. N. Engl. J. Med. 1969, 280, 787–798.
- O’Sullivan, J.A.; Bochner, B.S. Eosinophils and eosinophil-associated diseases: An update. J. Allergy Clin. Immunol. 2018, 141, 505–517.
- Yanagibashi, T.; Satoh, M.; Nagai, Y.; Koike, M.; Takatsu, K. Allergic diseases: From bench to clinic-Contribution of the discovery of interleukin-5. Cytokine 2017, 98, 59–70.
- Nair, P.; Pizzichini, M.M.; Kjarsgaard, M.; Inman, M.D.; Efthimiadis, A.; Pizzichini, E.; Hargreave, F.; O’Byrne, P.M. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N. Engl. J. Med. 2009, 360, 985–993.
- Haldar, P.; Brightling, C.E.; Hargadon, B.; Gupta, S.; Monteiro, W.; Sousa, A.; Marshall, R.P.; Bradding, P.; Green, R.H.; Wardlaw, A.J.; et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N. Engl. J. Med. 2009, 360, 973–984.
- Pavord, I.D.; Korn, S.; Howarth, P.; Bleecker, E.R.; Buhl, R.; Keene, O.N.; Ortega, H.; Chanez, P. Mepolizumab for severe eosinophilic asthma (DREAM): A multicentre, double-blind, placebo-controlled trial. Lancet 2012, 380, 651–659.
- Wechsler, M.E.; Akuthota, P.; Jayne, D.; Khoury, P.; Klion, A.; Langford, C.A.; Merkel, P.A.; Moosig, F.; Specks, U.; Cid, M.C.; et al. EGPA Mepolizumab Study Team. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. N. Engl. J. Med. 2017, 376, 1921–1932.
- Sarkis, E.; Patel, S.; Burns, K.; Batarseh, H.; Mador, M.J. Anti-interleukin (IL)-5 as a steroid-sparing agent in chronic eosinophilic pneumonia. J. Asthma. 2018, 57, 1–5.
- To, M.; Kono, Y.; Yamawaki, S.; Soeda, S.; Katsube, O.; Kishi, H.; To, Y. A case of chronic eosinophilic pneumonia successfully treated with mepolizumab. J. Allergy Clin. Immunol. Pract. 2018, 6, 1746–1748.
- Nakagome, K.; Nagata, M. Involvement and Possible Role of Eosinophils in Asthma Exacerbation. Front. Immunol. 2018, 18, 2220.
- Nakagome, K.; Nagata, M. Pathogenesis of airway inflammation in bronchial asthma. Auris. Nasus. Larynx. 2011, 38, 555–563.
- Nagata, M.; Sedgwick, J.B.; Bates, M.E.; Kita, H.; Busse, W.W. Eosinophil adhesion to vascular cell adhesion molecule-1 activates superoxide anion generation. J. Immunol. 1995, 155, 2194–2202.
- Nagata, M.; Sedgwick, J.B.; Kita, H.; Busse, W.W. Granulocyte macrophage colony-stimulating factor augments ICAM-1 and VCAM-1 activation of eosinophil function. Am. J. Respir. Cell Mol. Biol. 1998, 19, 158–166.
- Nagata, M.; Sedgwick, J.B.; Vrtis, R.; Busse, W.W. Endothelial cells upregulate eosinophil superoxide generation via VCAM-1 expression. Clin. Exp. Allergy. 1999, 29, 550–561.
- Nagata, M.; Yamamoto, H.; Tabe, K.; Sakamoto, Y. Eosinophil transmigration across VCAM-1-expressing endothelial cells is upregulated by antigen-stimulated mononuclear cells. Int. Arch. Allergy Immunol. 2001, 125 (Suppl 1), 7–11.
- Ying, S.; Robinson, D.S.; Meng, Q.; Rottman, J.; Kennedy, R.; Ringler, D.J.; Mackay, C.R.; Daugherty, B.L.; Springer, M.S.; Durham, S.R.; et al. Enhanced expression of eotaxin and CCR3 mRNA and protein in atopic asthma. Association with airway hyperresponsiveness and predominant co-localization of eotaxin mRNA to bronchial epithelial and endothelial cells. Eur. J. Immunol. 1997, 27, 3507–3516.
- Katoh, S.; Matsumoto, N.; Fukushima, K.; Mukae, H.; Kadota, J.I.; Kohno, S.; Matsukura, S. Elevated chemokine levels in bronchoalveolar lavage fluid of patients with eosinophilic pneumonia. J. Allergy Clin. Immunol. 2000, 106, 730–736.
- Tateno, H.; Nakamura, H.; Minematsu, N.; Amakawa, K.; Terashima, T.; Fujishima, S.; Luster, A.D.; Lilly, C.M.; Yamaguchi, K. Eotaxin and monocyte chemoattractant protein-1 in chronic eosinophilic pneumonia. Eur. Respir. J. 2001, 17, 962–968.
- Nakagome, K.; Shoda, H.; Shirai, T.; Nishihara, F.; Soma, T.; Uchida, Y.; Sakamoto, Y.; Nagata, M. Eosinophil transendothelial migration induced by the bronchoalveolar lavage fluid of acute eosinophilic pneumonia. Respirology 2017, 22, 913–921.
- Kobayashi, Y.; Konno, Y.; Kanda, A.; Yamada, Y.; Yasuba, H.; Sakata, Y.; Fukuchi, M.; Tomoda, K.; Iwai, H.; Ueki, S. Critical role of CCL4 in eosinophil recruitment into the airway. Clin. Exp. Allergy. 2019, 49, 853–860.
- Liu, L.Y.; Sedgwick, J.B.; Bates, M.E.; Vrtis, R.F.; Gern, J.E.; Kita, H.; Jarjour, N.N.; Busse, W.W.; Kelly, E.A. Decreased expression of membrane IL-5 receptor alpha on human eosinophils: I. Loss of membrane IL-5 receptor alpha on airway eosinophils and increased soluble IL-5 receptor alpha in the airway after allergen challenge. J. Immunol. 2002, 169, 6452–6458.
- Liu, L.Y.; Sedgwick, J.B.; Bates, M.E.; Vrtis, R.F.; Gern, J.E.; Kita, H.; Jarjour, N.N.; Busse, W.W.; Kelly, E.A. Decreased expression of membrane IL-5 receptor alpha on human eosinophils: II. IL-5 down-modulates its receptor via a proteinase-mediated process. J. Immunol. 2002, 169, 6459–6466.
- Nagata, M.; Saito, K.; Tsuchiya, T.; Sakamoto, Y. Leukotriene D4 upregulates eosinophil adhesion via the cysteinyl leukotriene 1 receptor. J. Allergy Clin. Immunol. 2002, 109, 676–680.
- Saito, K.; Nagata, M.; Kikuchi, I.; Sakamoto, Y. Leukotriene D4 and eosinophil transendothelial migration, superoxide generation, and degranulation via β2 integrin. Ann. Allergy Asthma Immunol. 2004, 93, 594–600.
- Kitaura, M.; Nakajima, T.; Imai, T.; Harada, S.; Combadiere, C.; Tiffany, H.L.; Murphy, P.M.; Yoshie, O. Molecular cloning of human eotaxin, an eosinophil-selective CC chemokine, and identification of a specific eosinophil eotaxin receptor, CC chemokine receptor 3. J. Biol. Chem. 1996, 271, 7725–7730.
- Nagase, H.; Miyamasu, M.; Yamaguchi, M.; Fujisawa, T.; Ohta, K.; Yamamoto, K.; Morita, Y.; Hirai, K. Expression of CXCR4 in eosinophils: Functional analyses and cytokine-mediated regulation. J. Immunol. 2000, 164, 5935–5943.
- Jinquan, T.; Jing, C.; Jacobi, H.H.; Reimert, C.M.; Millner, A.; Quan, S.; Hansen, J.B.; Dissing, S.; Malling, H.J.; Skov, P.S.; et al. CXCR3 expression and activation of eosinophils: Role of IFN-γ-inducible protein-10 and monokine induced by IFN-γ. J. Immunol. 2000, 165, 1548–1556.
- Wilkerson, E.M.; Johansson, M.W.; Hebert, A.S.; Westphall, M.S.; Mathur, S.K.; Jarjour, N.N.; Schwantes, E.A.; Mosher, D.F.; Coon, J.J. The Peripheral Blood Eosinophil Proteome. J. Proteome Res. 2016, 15, 1524–1533.
- Larose, M.C.; Archambault, A.S.; Provost, V.; Laviolette, M.; Flamand, N. Regulation of Eosinophil and Group 2 Innate Lymphoid Cell Trafficking in Asthma. Front Med. 2017, 4, 136.
- Fujisawa, T.; Kato, Y.; Atsuta, J.; Terada, A.; Iguchi, K.; Kamiya, H.; Yamada, H.; Nakajima, T.; Miyamasu, M.; Hirai, K. Chemokine production by the BEAS-2B human bronchial epithelial cells: Differential regulation of eotaxin, IL-8, and RANTES by TH2- and TH1-derived cytokines. J. Allergy Clin. Immunol. 2000, 105, 126–133.
- White, J.R.; Imburgia, C.; Dul, E.; Appelbaum, E.; O’Donnell, K.; O’Shannessy, D.J.; Brawner, M.; Fornwald, J.; Adamou, J.; Elshourbagy, N.A.; et al. Cloning and functional characterization of a novel human CC chemokine that binds to the CCR3 receptor and activates human eosinophils. J. Leukoc. Biol. 1997, 62, 667–675.
- Lamkhioued, B.; Garcia-Zepeda, E.A.; Abi-Younes, S.; Nakamura, H.; Jedrzkiewicz, S.; Wagner, L.; Renzi, P.M.; Allakhverdi, Z.; Lilly, C.; Hamid, Q.; et al. Monocyte chemoattractant protein (MCP)-4 expression in the airways of patients with asthma. Induction in epithelial cells and mononuclear cells by proinflammatory cytokines. Am. J. Respir. Crit. Care Med. 2000, 162, 723–732.
- Mueller, A.; Strange, P.G. The chemokine receptor, CCR5. Int. J. Biochem. Cell Biol. 2004, 36, 35–38.
- Wells, T.N.; Power, C.A.; Shaw, J.P.; Proudfoot, A.E. Chemokine blockers-therapeutics in the making? Trends Pharmacol. Sci. 2006, 27, 41–47.
- Fuchimoto, Y.; Kanehiro, A.; Miyahara, N.; Koga, H.; Ikeda, G.; Waseda, K.; Tanimoto, Y.; Ueha, S.; Kataoka, M.; Gelfand, E.W.; et al. Requirement for chemokine receptor 5 in the development of allergen-induced airway hyperresponsiveness and inflammation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1248–1255.
- Katoh, S.; Matsumoto, N.; Matsumoto, K.; Fukushima, K.; Matsukura, S. Elevated interleukin-18 levels in bronchoalveolar lavage fluid of patients with eosinophilic pneumonia. Allergy 2004, 59, 850–856.
- Miyazaki, E.; Nureki, S.; Fukami, T.; Shigenaga, T.; Ando, M.; Ito, K.; Ando, H.; Sugisaki, K.; Kumamoto, T.; Tsuda, T. Elevated levels of thymus- and activation-regulated chemokine in bronchoalveolar lavage fluid from patients with eosinophilic pneumonia. Am. J. Respir. Crit. Care Med. 2002, 165, 1125–1131.
- Mato, N.; Bando, M.; Kusano, A.; Hirano, T.; Nakayama, M.; Uto, T.; Nakaya, T.; Yamasawa, H.; Sugiyama, Y. Clinical significance of interleukin 33 (IL-33) in patients with eosinophilic pneumonia. Allergol. Int. 2013, 62, 45–52.
- Nakagome, K.; Nakamura, Y.; Kobayashi, T.; Ohta, S.; Ono, J.; Kobayashi, K.; Ikebuchi, K.; Noguchi, T.; Soma, T.; Yamauchi, K.; et al. Elevated Periostin Concentrations in the Bronchoalveolar Lavage Fluid of Patients with Eosinophilic Pneumonia. Int. Arch. Allergy Immunol. 2019, 178, 264–271.
- Castro, M.; Corren, J.; Pavord, I.D.; Maspero, J.; Wenzel, S.; Rabe, K.F.; Busse, W.W.; Ford, L.; Sher, L.; FitzGerald, J.M.; et al. Dupilumab Efficacy and Safety in Moderate-to-Severe Uncontrolled Asthma. N. Engl. J. Med. 2018, 378, 2486–2496.
- Bachert, C.; Han, J.K.; Desrosiers, M.; Hellings, P.W.; Amin, N.; Lee, S.E.; Mullol, J.; Greos, L.S.; Bosso, J.V.; Laidlaw, T.M.; et al. Efficacy and safety of dupilumab in patients with severe chronic rhinosinusitis with nasal polyps (LIBERTY NP SINUS-24 and LIBERTY NP SINUS-52): Results from two multicentre, randomised, double-blind, placebo-controlled, parallel-group phase 3 trials. Lancet 2019, 394, 1638–1650.

