 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natalia Yanguas Casás | + 3521 word(s) | 3521 | 2021-06-15 09:02:55 | | | |
| 2 | Peter Tang | -1 word(s) | 3520 | 2021-06-21 05:23:41 | | |
Video Upload Options
Lymphoma research is a paradigm of integrating basic and applied research within the fields of molecular marker-based diagnosis and therapy. In recent years, major advances in next-generation sequencing have substantially improved the understanding of the genomics underlying diffuse large B-cell lymphoma (DLBCL), the most frequent type of B-cell lymphoma. This review addresses the various approaches that have helped unveil the biology and intricate alterations in this pathology, from cell lines to more sophisticated last-generation experimental models, such as organoids. We also provide an overview of the most recent findings in the field, their potential relevance for designing targeted therapies and the corresponding applicability to personalized medicine.
1. Unraveling Aggressive B-Cell Lymphomas: The Elusive Link between Genomics and Personalized Treatment
2. Diffuse Large B-Cell Lymphomas: Clinically Unresolved Genomic Complexity
2.1. Diffuse Large B-Cell Lymphoma: A Traditional Overview
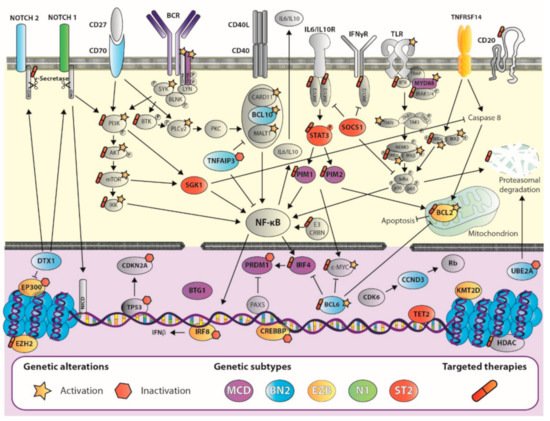
2.2. Advances in DLBCL Genomics: Genetic Signature Helps Design Tailored Therapies
2.3. Friend or Foe: The DLBCL Microenvironment
3. Functional Models for B-Cell Lymphoma Research
3.1. Gene Editing of Lymphoma Cells
3.2. The Rise of Drug Screening: Mission Pathways
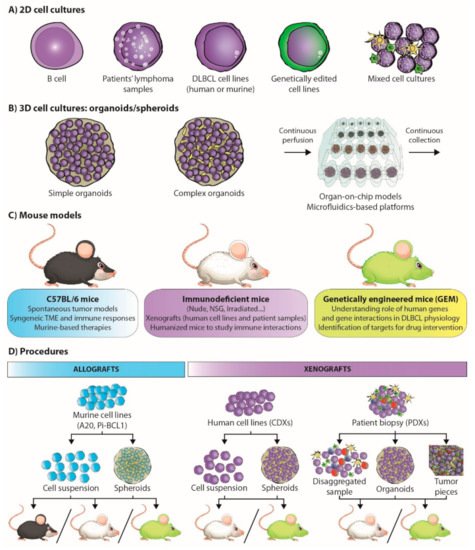
3.3. Of Mice and Men: Understanding Lymphoma Biology
|
Strategy |
Phenotype/Incidence |
Prospective Uses |
|
|---|---|---|---|
|
Genetically engineered mice |
Eμ-Myc |
DLBCL (time dependent) [70] |
|
|
Eμ-BRD2 |
DLBCL [61] |
||
|
Bcl6 Knock in |
GC-DLBCL [62] |
||
|
Bcl6/Myc |
ABC-DLBCL [62] |
||
|
Iµ:HA.BCL6 |
36–62% lymphoma incidence [62] |
Combination with conditional Spen and Tnfaip3 knockout or oncogenic Notch2 alleles to model Cluster BN2 |
|
|
Mb1:Cre;Eµ:Bcl2;Crebbpfl/fl |
GC-DLBCL [71] |
Combination of the different alleles to generate a sophisticated EZB mouse model |
|
|
Cγ1Cre/wt;Kmt2dfl/fl;VavP:Bcl2 |
21% incidence GC-DLBCL [72] |
||
|
Ezh2cond.p.Y641F/wt;VavP:Bcl2; Cγ1Cre/wt |
DLBCL-like lymphoma [73] |
||
|
Cd19Cre/wt;Myd88cond.p.L252P/wt;Rosa26LSL.BCL2-IRES-GFP/wt |
85% incidence ABC-DLBCL [74] |
Modeling of the MCD cluster by combination of both alleles and a newly generated CD79Bcond.p.Y196H/wt allele, or with the already existing Prdm1fl/fl |
|
|
Cγ1Cre;Prdm1fl/fl;Rosa26LSL.IKK2ca |
50% incidence IRF4, post-GC DLBCL [75] |
||
|
Syngeneic models |
Pi-BCL1 (m) iv or ip in BALB/c immunocompetent mice |
Gene editing Generation of complex organoids prior to inoculation of the cell line |
|
|
A20 (m) iv, intrasplenic, or sc injection in BALB/c immunocompetent mice |
|||
|
Xenograft models |
SU-DHL-4 (h) iv or sc in SCID immunodeficient mice |
Gene editing Generation of organoids Inoculation in humanized NOD/SCID mice |
|
|
Transduced HPCs in irradiated mice |
Generation of a-la-carte HPCs reproducing the genetic signatures Inoculation in humanized NOD/SCID mice |
||
|
PDX iv or sc in immunodeficient mice |
20–30% successful engraftment |
Use of humanized mice to study lymphoma physiology and drug responses Personalized medicine |
|
|
PDO iv or sc in immunodeficient mice |
20–30% successful engraftment |
Genetic modification Use of humanized mice to study lymphoma physiology and drug responses Personalized medicine. |
The headings of each mouse model subtype are highlighted in bold.
3.4. Back to the Bench: À La Carte-Engineered Spheroids
References
- Bakhshi, T.J.; Georgel, P.T. Genetic and Epigenetic Determinants of Diffuse Large B-Cell Lymphoma. Blood Cancer J. 2020, 10, 1–23.
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadié, M.; Simonetti, A.; Lutz, J.-M.; et al. Incidence of Hematologic Malignancies in Europe by Morphologic Subtype: Results of the HAEMACARE Project. Blood 2010, 116, 3724–3734.
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. Diffuse Large B-Cell Lymphoma—Cancer Stat Facts. 2019. Available online: (accessed on 18 May 2021).
- Sehn, L.H.; Salles, G. Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2021.
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profiling. Nature 2000, 403, 503–511.
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The Use of Molecular Profiling to Predict Survival after Chemotherapy for Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947.
- Jaffe, E.S.; Barr, P.M.; Smith, S.M. Understanding the New WHO Classification of Lymphoid Malignancies: Why It’s Important and How It Will Affect Practice. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 535–546.
- Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008; ISBN 978-92-832-4494-3.
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390.
- Basso, K.; Dalla-Favera, R. Germinal Centres and B Cell Lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172–184.
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the Coding Genome of Diffuse Large B-Cell Lymphoma. Nat. Genet. 2011, 43, 830.
- Dubois, S.; Viailly, P.-J.; Mareschal, S.; Bohers, E.; Bertrand, P.; Ruminy, P.; Maingonnat, C.; Jais, J.-P.; Peyrouze, P.; Figeac, M.; et al. Next-Generation Sequencing in Diffuse Large B-Cell Lymphoma Highlights Molecular Divergence and Therapeutic Opportunities: A LYSA Study. Clin. Cancer Res. 2016, 22, 2919–2928.
- Mandelbaum, J.; Bhagat, G.; Tang, H.; Mo, T.; Brahmachary, M.; Shen, Q.; Chadburn, A.; Rajewsky, K.; Tarakhovsky, A.; Pasqualucci, L.; et al. BLIMP1 Is a Tumor Suppressor Gene Frequently Disrupted in Activated B Cell-like Diffuse Large B Cell Lymphoma. Cancer Cell 2010, 18, 568–579.
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent Mutation of Histone-Modifying Genes in Non-Hodgkin Lymphoma. Nature 2011, 476, 298–303.
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic Active B-Cell-Receptor Signalling in Diffuse Large B-Cell Lymphoma. Nature 2010, 463, 88–92.
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of Multiple Genes Cause Deregulation of NF-KappaB in Diffuse Large B-Cell Lymphoma. Nature 2009, 459, 717–721.
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and Prioritization of Somatic Mutations in Diffuse Large B-Cell Lymphoma (DLBCL) by Whole-Exome Sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879.
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.A.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic Heterogeneity of Diffuse Large B-Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403.
- Novak, A.J.; Asmann, Y.W.; Maurer, M.J.; Wang, C.; Slager, S.L.; Hodge, L.S.; Manske, M.; Price-Troska, T.; Yang, Z.-Z.; Zimmermann, M.T.; et al. Whole-Exome Analysis Reveals Novel Somatic Genomic Alterations Associated with Outcome in Immunochemotherapy-Treated Diffuse Large B-Cell Lymphoma. Blood Cancer J. 2015, 5, e346.
- Karube, K.; Enjuanes, A.; Dlouhy, I.; Jares, P.; Martin-Garcia, D.; Nadeu, F.; Ordóñez, G.R.; Rovira, J.; Clot, G.; Royo, C.; et al. Integrating Genomic Alterations in Diffuse Large B-Cell Lymphoma Identifies New Relevant Pathways and Potential Therapeutic Targets. Leukemia 2018, 32, 675–684.
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically Active MYD88 Mutations in Human Lymphoma. Nature 2011, 470, 115–119.
- Monti, S.; Chapuy, B.; Takeyama, K.; Rodig, S.J.; Hao, Y.; Yeda, K.T.; Inguilizian, H.; Mermel, C.; Currie, T.; Dogan, A.; et al. Integrative Analysis Reveals an Outcome-Associated and Targetable Pattern of P53 and Cell Cycle Deregulation in Diffuse Large B Cell Lymphoma. Cancer Cell 2012, 22, 359–372.
- Jardin, F.; Jais, J.-P.; Molina, T.-J.; Parmentier, F.; Picquenot, J.-M.; Ruminy, P.; Tilly, H.; Bastard, C.; Salles, G.-A.; Feugier, P.; et al. Diffuse Large B-Cell Lymphomas with CDKN2A Deletion Have a Distinct Gene Expression Signature and a Poor Prognosis under R-CHOP Treatment: A GELA Study. Blood 2010, 116, 1092–1104.
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal Gene Signatures in Large-B-Cell Lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323.
- Challa-Malladi, M.; Lieu, Y.K.; Califano, O.; Holmes, A.B.; Bhagat, G.; Murty, V.V.; Dominguez-Sola, D.; Pasqualucci, L.; Dalla-Favera, R. Combined Genetic Inactivation of Β2-Microglobulin and CD58 Reveals Frequent Escape from Immune Recognition in Diffuse Large B Cell Lymphoma. Cancer Cell 2011, 20, 728–740.
- Miao, Y.; Medeiros, L.J.; Li, Y.; Li, J.; Young, K.H. Genetic Alterations and Their Clinical Implications in DLBCL. Nat. Rev. Clin. Oncol. 2019, 16, 634–652.
- Karube, K.; Campo, E. MYC Alterations in Diffuse Large B-Cell Lymphomas. Semin. Hematol. 2015, 52, 97–106.
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407.
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15.
- Pasqualucci, L.; Dalla-Favera, R. The Genetic Landscape of Diffuse Large B-Cell Lymphoma. Semin. Hematol. 2015, 52, 67–76.
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e14.
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular Subtypes of Diffuse Large B Cell Lymphoma Are Associated with Distinct Pathogenic Mechanisms and Outcomes. Nat. Med. 2018, 24, 679–690.
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted Sequencing in DLBCL, Molecular Subtypes, and Outcomes: A Haematological Malignancy Research Network Report. Blood 2020, 135, 1759–1771.
- Pedrosa, L.; Fernández-Miranda, I.; Pérez-Callejo, D.; Quero, C.; Rodríguez, M.; Martín-Acosta, P.; Gómez, S.; González-Rincón, J.; Santos, A.; Tarin, C.; et al. Proposal and Validation of a Method to Classify Genetic Subtypes of Diffuse Large B Cell Lymphoma. Sci. Rep. 2021, 11, 1886.
- Runge, H.F.P.; Lacy, S.; Barrans, S.; Beer, P.A.; Painter, D.; Smith, A.; Roman, E.; Burton, C.; Crouch, S.; Tooze, R.; et al. Application of the LymphGen Classification Tool to 928 Clinically and Genetically-Characterised Cases of Diffuse Large B Cell Lymphoma (DLBCL). Br. J. Haematol. 2021, 192, 216–220.
- Young, R.M.; Phelan, J.D.; Wilson, W.H.; Staudt, L.M. Pathogenic B-Cell Receptor Signaling in Lymphoid Malignancies: New Insights to Improve Treatment. Immunol. Rev. 2019, 291, 190–213.
- Young, R.M.; Phelan, J.D.; Shaffer, A.L.; Wright, G.W.; Huang, D.W.; Schmitz, R.; Johnson, C.; Oellerich, T.; Wilson, W.; Staudt, L.M. Taming the Heterogeneity of Aggressive Lymphomas for Precision Therapy. Annu. Rev. Cancer Biol. 2019, 3, 429–455.
- Ciavarella, S.; Vegliante, M.C.; Fabbri, M.; De Summa, S.; Melle, F.; Motta, G.; De Iuliis, V.; Opinto, G.; Enjuanes, A.; Rega, S.; et al. Dissection of DLBCL Microenvironment Provides a Gene Expression-Based Predictor of Survival Applicable to Formalin-Fixed Paraffin-Embedded Tissue. Ann. Oncol. 2018, 29, 2363–2370.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674.
- Scott, D.W.; Gascoyne, R.D. The Tumour Microenvironment in B Cell Lymphomas. Nat. Rev. Cancer 2014, 14, 517–534.
- Kotlov, N.; Bagaev, A.; Revuelta, M.V.; Phillip, J.M.; Cacciapuoti, M.T.; Antysheva, Z.; Svekolkin, V.; Tikhonova, E.; Miheecheva, N.; Kuzkina, N.; et al. Clinical and Biological Subtypes of B-Cell Lymphoma Revealed by Microenvironmental Signatures. Cancer Discov. 2021.
- Clozel, T.; Yang, S.; Elstrom, R.L.; Tam, W.; Martin, P.; Kormaksson, M.; Banerjee, S.; Vasanthakumar, A.; Culjkovic, B.; Scott, D.W.; et al. Mechanism-Based Epigenetic Chemosensitization Therapy of Diffuse Large B-Cell Lymphoma. Cancer Discov. 2013, 3, 1002–1019.
- Mulder, T.A.; Wahlin, B.E.; Österborg, A.; Palma, M. Targeting the Immune Microenvironment in Lymphomas of B-Cell Origin: From Biology to Clinical Application. Cancers 2019, 11, 915.
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of A•T to G•C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471.
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic Genome Editing: Prospects and Challenges. Nat. Med. 2015, 21, 121–131.
- Caeser, R.; Di Re, M.; Krupka, J.A.; Gao, J.; Lara-Chica, M.; Dias, J.M.L.; Cooke, S.L.; Fenner, R.; Usheva, Z.; Runge, H.F.P.; et al. Genetic Modification of Primary Human B Cells to Model High-Grade Lymphoma. Nat. Commun. 2019, 10, 4543.
- Bai, B.; Myklebust, J.H.; Wälchli, S. Gene Editing in B-Lymphoma Cell Lines Using CRISPR/Cas9 Technology. Methods Mol. Biol. 2020, 2115, 445–454.
- Felce, S.L.; Anderson, A.P.; Maguire, S.; Gascoyne, D.M.; Armstrong, R.N.; Wong, K.K.; Li, D.; Banham, A.H. CRISPR/Cas9-Mediated Foxp1 Silencing Restores Immune Surveillance in an Immunocompetent A20 Lymphoma Model. Front. Oncol. 2020, 10, 448.
- Greiner, V.; Puerto, R.B.; Liu, S.; Herbel, C.; Carmona, E.M.; Goldberg, M.S. CRISPR-Mediated Editing of the B Cell Receptor in Primary Human B Cells. iScience 2019, 12, 369–378.
- Schlager, S.; Salomon, C.; Olt, S.; Albrecht, C.; Ebert, A.; Bergner, O.; Wachter, J.; Trapani, F.; Gerlach, D.; Voss, T.; et al. Inducible Knock-out of BCL6 in Lymphoma Cells Results in Tumor Stasis. Oncotarget 2020, 11, 875–890.
- Nie, M.; Du, L.; Zhang, B.; Ren, W.; Joung, J.; Ye, X.; Fuenzalida, J.A.; Shi, X.; Liu, D.; Wu, K.; et al. Essentiality of CREBBP in EP300 Truncated B-Cell Lymphoma Revealed by Genome-Wide CRISPR-Cas9 Screen. bioRxiv 2019, 746594.
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117.
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607.
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic Identification of Genomic Markers of Drug Sensitivity in Cancer Cells. Nature 2012, 483, 570–575.
- Haverty, P.M.; Lin, E.; Tan, J.; Yu, Y.; Lam, B.; Lianoglou, S.; Neve, R.M.; Martin, S.; Settleman, J.; Yauch, R.L.; et al. Reproducible Pharmacogenomic Profiling of Cancer Cell Line Panels. Nature 2016, 533, 333–337.
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508.
- Quentmeier, H.; Pommerenke, C.; Dirks, W.G.; Eberth, S.; Koeppel, M.; MacLeod, R.A.F.; Nagel, S.; Steube, K.; Uphoff, C.C.; Drexler, H.G. The LL-100 Panel: 100 Cell Lines for Blood Cancer Studies. Sci. Rep. 2019, 9, 8218.
- Kohnken, R.; Porcu, P.; Mishra, A. Overview of the Use of Murine Models in Leukemia and Lymphoma Research. Front. Oncol. 2017, 7, 22.
- Perlman, R.L. Mouse Models of Human Disease: An Evolutionary Perspective. Evol. Med. Public Health 2016, 2016, 170–176.
- Harris, A.W.; Pinkert, C.A.; Crawford, M.; Langdon, W.Y.; Brinster, R.L.; Adams, J.M. The E Mu-Myc Transgenic Mouse. A Model for High-Incidence Spontaneous Lymphoma and Leukemia of Early B Cells. J. Exp. Med. 1988, 167, 353–371.
- Greenwald, R.J.; Tumang, J.R.; Sinha, A.; Currier, N.; Cardiff, R.D.; Rothstein, T.L.; Faller, D.V.; Denis, G.V. E Mu-BRD2 Transgenic Mice Develop B-Cell Lymphoma and Leukemia. Blood 2004, 103, 1475–1484.
- Cattoretti, G.; Pasqualucci, L.; Ballon, G.; Tam, W.; Nandula, S.V.; Shen, Q.; Mo, T.; Murty, V.V.; Dalla-Favera, R. Deregulated BCL6 Expression Recapitulates the Pathogenesis of Human Diffuse Large B Cell Lymphomas in Mice. Cancer Cell 2005, 7, 445–455.
- Cho, S.-Y.; Kang, W.; Han, J.Y.; Min, S.; Kang, J.; Lee, A.; Kwon, J.Y.; Lee, C.; Park, H. An Integrative Approach to Precision Cancer Medicine Using Patient-Derived Xenografts. Mol. Cells 2016, 39, 77–86.
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L.; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A Multiprotein Supercomplex Controlling Oncogenic Signalling in Lymphoma. Nature 2018, 560, 387–391.
- Abdelrasoul, H.; Werner, M.; Setz, C.S.; Okkenhaug, K.; Jumaa, H. PI3K Induces B-Cell Development and Regulates B Cell Identity. Sci. Rep. 2018, 8, 1327.
- Béguelin, W.; Teater, M.; Meydan, C.; Hoehn, K.B.; Phillip, J.M.; Soshnev, A.A.; Venturutti, L.; Rivas, M.A.; Calvo-Fernández, M.T.; Gutierrez, J.; et al. Mutant EZH2 Induces a Pre-Malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell 2020, 37, 655–673.e11.
- Brescia, P.; Schneider, C.; Holmes, A.B.; Shen, Q.; Hussein, S.; Pasqualucci, L.; Basso, K.; Dalla-Favera, R. MEF2B Instructs Germinal Center Development and Acts as an Oncogene in B Cell Lymphomagenesis. Cancer Cell 2018, 34, 453–465.e9.
- Dominguez, P.M.; Ghamlouch, H.; Rosikiewicz, W.; Kumar, P.; Béguelin, W.; Fontán, L.; Rivas, M.A.; Pawlikowska, P.; Armand, M.; Mouly, E.; et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-Cell Lymphomagenesis. Cancer Discov. 2018, 8, 1632–1653.
- Zhang, J.; Vlasevska, S.; Wells, V.A.; Nataraj, S.; Holmes, A.B.; Duval, R.; Meyer, S.N.; Mo, T.; Basso, K.; Brindle, P.K.; et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-Cell Lymphoma. Cancer Discov. 2017, 7, 322–337.
- Mori, S.; Rempel, R.E.; Chang, J.T.; Yao, G.; Lagoo, A.S.; Potti, A.; Bild, A.; Nevins, J.R. Utilization of Pathway Signatures to Reveal Distinct Types of B Lymphoma in the Emicro-Myc Model and Human Diffuse Large B-Cell Lymphoma. Cancer Res. 2008, 68, 8525–8534.
- García-Ramírez, I.; Tadros, S.; González-Herrero, I.; Martín-Lorenzo, A.; Rodríguez-Hernández, G.; Moore, D.; Ruiz-Roca, L.; Blanco, O.; Alonso-López, D.; Rivas, J.D.L.; et al. Crebbp Loss Cooperates with Bcl2 Overexpression to Promote Lymphoma in Mice. Blood 2017, 129, 2645–2656.
- Zhang, J.; Dominguez-Sola, D.; Hussein, S.; Lee, J.-E.; Holmes, A.B.; Bansal, M.; Vlasevska, S.; Mo, T.; Tang, H.; Basso, K.; et al. Disruption of KMT2D Perturbs Germinal Center B Cell Development and Promotes Lymphomagenesis. Nat. Med. 2015, 21, 1190–1198.
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell 2013, 23, 677–692.
- Knittel, G.; Liedgens, P.; Korovkina, D.; Seeger, J.M.; Al-Baldawi, Y.; Al-Maarri, M.; Fritz, C.; Vlantis, K.; Bezhanova, S.; Scheel, A.H.; et al. B-Cell-Specific Conditional Expression of Myd88p.L252P Leads to the Development of Diffuse Large B-Cell Lymphoma in Mice. Blood 2016, 127, 2732–2741.
- Calado, D.P.; Zhang, B.; Srinivasan, L.; Sasaki, Y.; Seagal, J.; Unitt, C.; Rodig, S.; Kutok, J.; Tarakhovsky, A.; Schmidt-Supprian, M.; et al. Constitutive Canonical NF-ΚB Activation Cooperates with Disruption of BLIMP1 in the Pathogenesis of Activated B-Cell like Diffuse Large B-Cell Lymphoma. Cancer Cell 2010, 18, 580–589.
- Illidge, T.; Honeychurch, J.; Howatt, W.; Ross, F.; Wilkins, B.; Cragg, M. A New in Vivo and in Vitro B Cell Lymphoma Model, Pi-BCL1. Cancer Biother. Radiopharm. 2000, 15, 571–580.
- Timmerman, J.M.; Caspar, C.B.; Lambert, S.L.; Syrengelas, A.D.; Levy, R. Idiotype-Encoding Recombinant Adenoviruses Provide Protective Immunity against Murine B-Cell Lymphomas. Blood 2001, 97, 1370–1377.
- Chaise, C.; Itti, E.; Petegnief, Y.; Wirquin, E.; Copie-Bergman, C.; Farcet, J.-P.; Delfau-Larue, M.-H.; Meignan, M.; Talbot, J.-N.; Molinier-Frenkel, V. [F-18]-Fluoro-2-Deoxy-D: -Glucose Positron Emission Tomography as a Tool for Early Detection of Immunotherapy Response in a Murine B Cell Lymphoma Model. Cancer Immunol. Immunother. 2007, 56, 1163–1171.
- Palmieri, C.; Falcone, C.; Iaccino, E.; Tuccillo, F.M.; Gaspari, M.; Trimboli, F.; De Laurentiis, A.; Luberto, L.; Pontoriero, M.; Pisano, A.; et al. In Vivo Targeting and Growth Inhibition of the A20 Murine B-Cell Lymphoma by an Idiotype-Specific Peptide Binder. Blood 2010, 116, 226–238.
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of Tryptophan Catabolism by Human Leukemic Cells Results in the Conversion of CD25- into CD25+ T Regulatory Cells. Blood 2007, 109, 2871–2877.
- Yan, J.-S.; Chen, X.-Y.; Li, W.-P.; Yang, Y.; Song, Z.-L. Establishing SCID Mouse Models of B-Cell Non-Hodgkin’s Lymphoma. Ai Zheng 2009, 28, 181–183.
- Ackler, S.; Xiao, Y.; Mitten, M.J.; Foster, K.; Oleksijew, A.; Refici, M.; Schlessinger, S.; Wang, B.; Chemburkar, S.R.; Bauch, J.; et al. ABT-263 and Rapamycin Act Cooperatively to Kill Lymphoma Cells In Vitro and In Vivo. Mol. Cancer Ther. 2008, 7, 3265–3274.
- Jiang, Y.; Ortega-Molina, A.; Geng, H.; Ying, H.-Y.; Hatzi, K.; Parsa, S.; McNally, D.; Wang, L.; Doane, A.S.; Agirre, X.; et al. CREBBP Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discov. 2017, 7, 38–53.
- Alizadeh, A.A.; Gentles, A.J.; Alencar, A.J.; Liu, C.L.; Kohrt, H.E.; Houot, R.; Goldstein, M.J.; Zhao, S.; Natkunam, Y.; Advani, R.H.; et al. Prediction of Survival in Diffuse Large B-Cell Lymphoma Based on the Expression of 2 Genes Reflecting Tumor and Microenvironment. Blood 2011, 118, 1350–1358.
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The Prognostic Landscape of Genes and Infiltrating Immune Cells across Human Cancers. Nat. Med. 2015, 21, 938–945.
- Pizzi, M.; Boi, M.; Bertoni, F.; Inghirami, G. Emerging Therapies Provide New Opportunities to Reshape the Multifaceted Interactions between the Immune System and Lymphoma Cells. Leukemia 2016, 30, 1805–1815.
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D Cell Culture—A Comparison of Different Types of Cancer Cell Cultures. Arch. Med. Sci. 2018, 14, 910–919.
- Kim, J.W.; Ho, W.J.; Wu, B.M. The Role of the 3D Environment in Hypoxia-Induced Drug and Apoptosis Resistance. Anticancer Res. 2011, 31, 3237–3245.
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016, 4, 12.
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-Chips at the Frontiers of Drug Discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260.
- Brassard, J.A.; Lutolf, M.P. Engineering Stem Cell Self-Organization to Build Better Organoids. Cell Stem Cell 2019, 24, 860–876.
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15.

