Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Meng Yang | + 2408 word(s) | 2408 | 2021-06-03 08:39:56 | | | |
| 2 | Lily Guo | + 184 word(s) | 2592 | 2021-06-24 03:35:38 | | | | |
| 3 | Conner Chen | Meta information modification | 2592 | 2021-10-12 06:00:01 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yang, M. NAFLD. Encyclopedia. Available online: https://encyclopedia.pub/entry/10998 (accessed on 21 June 2026).
Yang M. NAFLD. Encyclopedia. Available at: https://encyclopedia.pub/entry/10998. Accessed June 21, 2026.
Yang, Meng. "NAFLD" Encyclopedia, https://encyclopedia.pub/entry/10998 (accessed June 21, 2026).
Yang, M. (2021, June 18). NAFLD. In Encyclopedia. https://encyclopedia.pub/entry/10998
Yang, Meng. "NAFLD." Encyclopedia. Web. 18 June, 2021.
Copy Citation
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease and may progress to cirrhosis or even hepatocellular carcinoma. A number of steroid hormones are important regulators of lipid homeostasis through fine tuning the expression of genes related to lipid synthesis, export, and metabolism. Dysregulation of such pathways has been implicated in the pathogenesis of NAFLD.
nonalcoholic fatty liver disease
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) has been increasingly recognized as a worldwide health issue and is estimated to affect 20–30% of the general population [1]. It is a progressive disease ranging from steatosis to nonalcoholic steatohepatitis (NASH), which is a risk for the development of cirrhosis and hepatocellular carcinoma (HCC) [2][3]. Most patients with NAFLD have simple steatosis in the absence of hepatocyte injury. However, approximately 10–30% of patients with NAFLD develop NASH characterized by inflammation and fibrosis [4]. Over the last decade, it has been shown that NAFLD is a multisystem disease, affecting several extrahepatic organs and regulatory pathways [5]. NAFLD is tightly associated with obesity, metabolic syndrome, and type 2 diabetes [6]. All of these complications related to NAFLD pose significant health and economic burdens on patients and society.
The pathogenesis of NAFLD is complex and described by a “multiple-hit” hypothesis, which has been proposed to supersede the outdated “two-hit” hypothesis [7]. The “multiple-hit” hypothesis highlights the importance of genetic and epigenetic factors, nutritional factors, intestinal microbiota, insulin resistance, and hormones secreted from adipose tissue [7][8]. All forms of NAFLD tightly correlate with hepatic and peripheral insulin resistance in epidemiological, experimental, and human studies [9][10][11]. Insulin resistance upregulates de novo lipogenesis, inhibits β-oxidation of free fatty acids (FFAs) and increases the degradation of very-low-density lipoprotein (VLDL) in the liver, leading to an increase in hepatic fat accumulation [12][13][14]. It also promotes lipolysis rates in adipose tissue, leading to more fatty-acid efflux to the liver [15]. The intestinal microbiota has been found to alter bile acid metabolism to affect hepatic lipid handling and alter host immunity contributing to NASH [16]. There is increasing evidence reporting adiponectin as an adipokine with anti-inflammatory and antifibrogenic activity protecting liver parenchyma against steatosis and apoptosis [17].
Lipid accumulation within hepatocytes caused by these risk factors leads to steatosis, defined as the presence of fat comprising 5% of liver weight [18]. Exaggerated lipid accumulation can cause toxicity via diverse mechanisms. For instance, toxic lipid metabolites derived from FFAs impose oxidative stress on hepatocytes [19]. Furthermore, oxidative stress affects the function of the endoplasmic reticulum and mitochondria, as well as contributes to hepatic inflammation and fibrosis [20].
Steroid hormones have been discovered to play important roles in lipid metabolism. Deregulation of steroid hormone level or bioactivity has been implicated in various degree of hepatocellular damages. In Cushing’s syndrome, the high circulating steroid hormone (glucocorticoid) level causes visceral obesity, insulin resistance, and hepatic steatosis [21]. Activation of the renin–angiotensin–aldosterone system modulated by multiple steroid hormones and corresponding receptors causes liver inflammation and fibrosis [22][23]. Moreover, steroid hormone receptor-regulated target genes are involved in both cholesterol and fatty-acid metabolism [24] and, therefore, have been implicated in the pathogenesis of NAFLD.
2. Role of Steroid Hormones in Hepatic Steatosis and Metabolism
Lipid homeostasis is fine-tuned through four major pathways: uptake of circulating lipids, de novo lipogenesis, fatty-acid oxidation, and export of lipids in VLDL [25]. Hepatic steatosis occurs when there is an imbalance between lipid acquisition and lipid disposal. Increasing data have shown that steroid hormones are involved in hepatic steatosis by modulating these processes (Figure 1). We summarize steroid hormone-mediated effects of hepatic steatosis in animal models in Table 1.
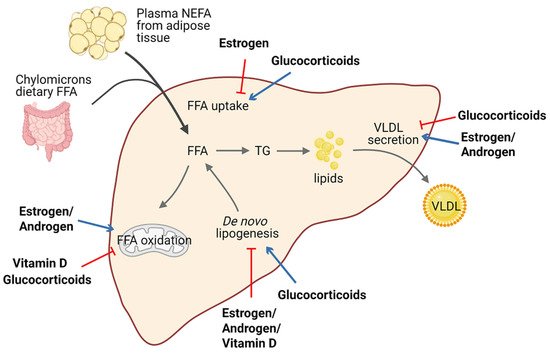
Figure 1. Schematic view of the roles of steroid hormones in hepatic lipid homeostasis. Steroid hormones have the ability to regulate lipid homeostasis via alteration of multiple major metabolic fates of hepatic FFAs. Blue arrows indicate stimulatory activity and red arrows indicate inhibitory activity. Abbreviations: free fatty acid (FFA); non-estesterified fatty acid (NEFA); triglyceride (TG); very-low-density lipoprotein (VLDL).
It is well established that estrogen and ERs are involved in the pathogenesis of NAFLD. For example, ERα-deficient mice have disrupted lipid metabolism, such as decreased fatty-acid oxidation and increased de novo lipogenesis, leading to elevated lipid accumulation in the liver compared with normal mice [26][27][28]. Similarly, downstream blocking of ERα contributed to higher visceral fat accumulation and reduced energy expenditure in female mice [29][26][30]. Meanwhile, E2 treatment promoted fatty-acid oxidation in the liver by increasing the expression of the fatty-acid transport protein, carnitine palmitoyltransferase 1 (CPT-1) [29]. In ovariectomized (OVX) female mice, estrogen replacement improves insulin sensitivity and facilitates the VLDL-mediated export of lipids from the liver by increasing hepatic VLDL-TG production [31]. Interestingly, long-term therapy of tamoxifen, a selective estrogen receptor modulator, can increase the risk of NAFLD in breast cancer patients [32].
Table 1. Summary of steroid hormones effects on hepatic steatosis. ↑ increased; ↓ decreased.
| Steroid Hormones | Model(s) Used | Major Phenotypes Examined |
|---|---|---|
| Estrogen | - Female ERα-deficient mice fed HFD for 10 weeks [27] - Male hepatic ERα-deficient mice fed HFD for 12 weeks [28] - OVX mice treated with E2 and fed HFD for 6 weeks [31] |
- Liver weight, hepatic steatosis, and ALT level ↑ - Hepatic steatosis and insulin resistance ↑ - Hepatic steatosis and insulin- mediated suppression of VLDL secretion ↓ |
| Androgen | - Male hepatic AR-deficient mice fed HFD for 8 weeks [33] |
- Body weight, hepatic steatosis, and insulin resistance ↑ |
| Glucocorticoid | - db/db mice treated with GC shRNA for 14 days [34] - SD rats treated with exogenous corticosterone and fed HFD for 16 days [35] |
- Hepatic steatosis and genes critical for lipid storage and transport ↓ - Hepatic steatosis, uptake of FA into liver, and ALT level ↑ |
| Mineralocorticoid | - Myeloid MR-deficient ob/ob mice [36] - Aldosterone synthase-deficient mice fed HFD for 12 weeks [37] |
- Hepatic steatosis, lipogenesis, and insulin resistance ↓ - HFD-feeding-induced hepatic steatosis ↓ |
| Vitamin D | - C57BL6 mice fed a high-fat/ high-sucrose diet followed by treatment with vitamin D for 15 weeks [38] - SD rats fed HFD followed by treatment with vitamin D for 16 weeks [39] |
- Hepatic steatosis and hepatic de novo lipogenesis ↓ - Liver weight, hepatic steatosis, and ALT level ↓ |
A meta-analysis revealed that lower serum testosterone levels are associated with men with NAFLD [40][41]. Testosterone deficiency in men displayed an increased accumulation of visceral adipose tissue and insulin resistance, which are factors favoring the development of hepatic steatosis [42]. Similar results were confirmed in hepatic AR-deficient mice, indicating that AR might play a role in the suppression of NAFLD [33][43]. Indeed, androgen/AR signaling was revealed to suppress fatty-acid synthesis by decreasing the expression of sterol regulatory element-binding protein (SREBP) and to induce insulin sensitivity by modulating phosphoinositide-3 kinase activity [24].
Conversely, glucocorticoids drive the expression of lipogenic genes including fatty-acid synthase (FASN) and acetyl-coA carboxylase 1 (Acaca) [44], stimulate de novo lipogenesis, and block VLDL secretion, thus resulting in hepatic steatosis [45]. Corticosterone, administered to rodent models to increase basal glucocorticoids levels, also induces an increase in Cluster Determinant 36 (CD36) expression that facilitates fatty-acid uptake, favoring the progression of hepatic steatosis [35]. Of note, patients with glucocorticoid excess may develop hepatic steatosis, as well as obesity and insulin resistance, in a significant proportion of cases [46][47]. Moreover, fatty liver development also represents a typical side-effect of long-term systemic glucocorticoids treatment during anti-inflammatory therapy. Additionally, mice deficient in hepatic GR displayed lower hepatic TG content and elevated levels of ketone bodies in the serum, indicating that disruption of GR expression reduces steatosis in db/db animals partly by triggering hepatic fatty-acid oxidation and ketogenesis [34].
Macrophage-specific deficiency of MR protects mice from hepatic steatosis and insulin resistance through the ERα/HGF/Met pathway [36]. In addition, aldosterone deficiency attenuates high-fat feeding-induced hepatic steatosis, although the underlying mechanism remains elusive [37]. On the other hand, elevated aldosterone level seems to enhance hepatic FFAs via modulation of lipogenesis and lipolysis [48].
Vitamin D has been proven to protect against high-fat diet-induced fatty liver by acting on the expression of de novo lipogenesis-related genes, including FASN and Acaca, and fatty-acid oxidation-related genes such as the acetyl-coA oxidase (Acox) [38][39]. In humans, hepatic VDR expression is inversely correlated with steatosis severity [49]. Activation of VDR in hepatic macrophages via its cognate ligand has been uncovered to ameliorate steatosis and insulin resistance in experimental studies [50].
3f. Role of Steroid Hormones in Hepatic Inflammation and Fibrosis
Accumulation of toxic lipids leads to ROS, endoplasmic reticulum (ER) stress, cell death, release of damage-associated molecular patterns (DAMPs), and secretion of inflammatory mediators and extracellular vesicles, which stimulate an inflammatory response in Kupffer cells and a fibrotic response in hepatic stellate cells (HSC) [51][52]. The inflammatory process includes recruitment of macrophages and neutrophils to the site of injured tissue, followed by production of proinflammatory chemokines and cytokines such as interleukin 1 (IL-1) and tumor necrosis factor alpha (TNF-α) [53][54]. Furthermore, initiation of the inflammatory acute-phase response of the liver is induced by these cytokines together with IL-6 [55]. Liver inflammation can lead to fibrosis that may eventually induce cirrhosis [56]. Hepatic fibrosis is characterized by the accumulation of extracellular matrix. As a crucial driver of fibrosis, HSCs can be transdifferentiated into proliferative and fibrogenic myofibroblasts [57]. Profibrogenic cytokines such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and transforming growth factor-β (TGF-β) are involved in the activation of HSCs and hepatic abnormal wound repair response [58][59][60]. Collectively, hepatocellular damage, inflammation, and fibrosis are the main characteristics of NASH [61][62]. Numerous studies have demonstrated the important effects of steroid hormones in the pathological progression of NASH (Figure 2). Here, we summarize steroid hormone-mediated effects on hepatic inflammation and fibrosis in animal models in Table 2.
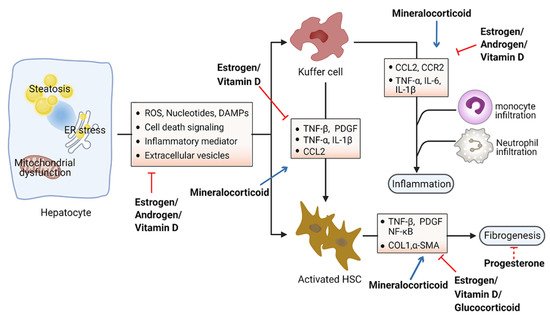
Figure 2. Schematic view of the roles of steroid hormones in NASH development. Steroid hormones are involved in guarding against hepatocellular damage, inflammation, and fibrosis in the pathological progression of NASH. Blue arrows indicate promoting activity and red arrows indicate inhibitory activity. Abbreviations: reactive oxygen species (ROS); damage-associated molecular patterns (DAMPs); tumor necrosis factor (TNF); platelet-derived growth factor (PDGF); interleukin (IL); C–C motif chemokine ligand 2 (CCL2); C–C motif chemokine receptor 2 (CCR2); nuclear factor kappa B (NK-kB); collagen I (COL1); α-smooth muscle actin (α-SMA).
Steroid hormones primarily display anti-inflammation and antifibrosis properties in the context of metabolic disorders. Estrogens have been found to directly suppress inflammatory processes in the liver. Women after menopause have decreased fatty-acid oxidation and increased lipogenesis in the liver, leading to the excessive accumulation of hepatic fat and culminating with inflammation [63][64]. Moreover, the generation of ROS and proinflammation cytokines was prevented by estrogen signaling in hepatocytes [65][66]. Estradiol treatment reduces hepatic inflammation in the animal model of NASH induced by methionine and a choline-deficient diet [67]. Furthermore, female mice with estrogen deficiency induced by OVX operation were observed with enhanced levels of macrophages infiltration and proinflammatory cytokines in the liver, including TNF-α, IL-6, C–C motif chemokine ligand 2 (CCL2), and C–C motif chemokine receptor 2 (CCR2) [68]. Hepatic fibrosis has also been shown to be associated with estrogen level. The risk of liver fibrosis increased significantly in a zebrafish experiment with ovarian senescence [69]. Similarly, a long duration of estrogen deficiency poses a high risk of hepatic fibrosis among postmenopausal women with NAFLD [70]. However, postmenopausal estrogen therapy may be considered for preventing the occurrence of liver fibrosis [71]. OVX female mice fed with a high-fat and high-cholesterol diet had obvious liver fibrosis with upregulated expression of collagen I α1, which could be improved by estrogen replacement therapy [66]. Additionally, β-LGND2, an ER-β-selective agonist, partially prevented inflammatory cell infiltration and liver fibrosis, providing a therapeutic benefit in NASH [72].
It has been shown that testosterone may increase the level of anti-inflammatory cytokines and reduce the level of proinflammatory cytokines, thereby exerting anti-inflammatory properties in patients with diabetes mellitus or coronary artery disease [73]. In experimental animal models of NAFLD, androgens were shown to be able to prevent NASH progression by acting on proinflammatory cytokines such as TNF-α and IL-6 [74].
Although increasing levels of progesterone have been associated with the development of systemic insulin resistance [75], little is known regarding the role of serum progesterone in NAFLD. Early studies have shown that treatment with progesterone in rabbits exerts a protective effect on hepatocytes against vacuolization and inflammation, accompanied by a low level of fibrosis [76].
Glucocorticoid hormones are considered the most widely used anti-inflammatory drugs. However, the effects of glucocorticoids on hepatic inflammation are still unclear, particularly in the progression of NAFLD. One study suggested that glucocorticoids may suppress the development of liver inflammation, considering that the perivascular infiltration of small mononuclear cells was diminished in the liver of mice following treatment with dexamethasone, a synthetic GR ligand [77]. Meanwhile, glucocorticoids suppress fibrotic gene expression including collagen I α1/2 by activating GR, which differentially regulates liver injury and hepatic fibrosis in HSCs or immune cells [77].
Table 2. Summary of the effects of steroid hormones on hepatic inflammation and fibrosis. ↑ increased; ↓ decreased.
| Steroid Hormones | Model (s) Used | Major Phenotypes Examined |
|---|---|---|
| Estrogen | - OVX mice fed HFD and high-fructose water for 12 weeks [68] - OVX mice fed a high-fat and high-cholic-acid diet for 6 weeks [66] - Old female zebrafish fed a high-calorie diet for 24 weeks [69] - Orchidectomized C57/BL6 mice treated with estradiol benzoate-fed MCD for 4 weeks [67] - Male C57BL6 mice treated with β-LGND2 and fed HFD for 10 weeks [72] |
- Hepatic inflammation and fibrosis, ALT level and ballooning degeneration ↑ - Liver fibrosis, inflammation, and hepatocyte ballooning degeneration ↑ - Liver fibrosis, IL-6, and TNF-β ↑ - Hepatic inflammation, MyD88, and IL-6 ↓ - Hepatic steatosis and insulin resistance ↓ |
| Androgen | - Orchidectomized male SD rats treated with dihydrotestosterone and fed HFD for 75 days [74] | - Portal inflammation, TNF-α, and IL-6 ↓ |
| Progesterone | - Hepatic fibrosis model of New Zealand male rabbits treated with progesterone for 180 days [76] | - Liver fibrosis, fat metamorphosis, and inflammatory infiltrate ↓ |
| Glucocorticoid | - Immune cell-specific GR-knockout mice treated with CCl4 and dexamethasone for 6 weeks [77] - HSC-specific GR-knockout mice treated with CCl4 and dexamethasone for 6 weeks [77] |
- Inflammation and monocyte recruitment ↓ - Hepatic fibrosis and fibrotic gene expression ↓ |
| Mineralocorticoid | - C57BL6 mice fed HFFD mixed with eplerenone for 12 weeks [78] - Male C57BL6 mice fed a CDAA diet for 22 weeks with eplerenone [79] - Male SD rats treated with aldosterone for 4 weeks [80] |
- Lipid accumulation, lobular inflammation, and collagen deposition ↓ - Hepatic fibrosis, steatosis, and inflammation ↓ - Hepatic fibrosis, oxidative stress, and DNA double-strand breaks ↑ |
| Vitamin D | - Vitamin D-deficient SD rats fed WD for 10 weeks [81] - CDAA diet-induced rat NASH model with phototherapy for 6 or 12 weeks [82] |
- Foci of lobular inflammation and ballooning degeneration ↑ - Collagen fibrosis, insulin and leptin resistance, inflammation, and HSC activation ↓ |
Aldosterone was observed to promote liver inflammation. ROS production can be stimulated by aldosterone in several tissues [83]. Blockade of aldosterone interaction with MR by eplerenone impeded macrophage infiltration and suppressed the expression of TNF-α and multiple copies in T-cell lymphoma-1 (MCT-1) in Kupffer cells, consequently ameliorating the development of NASH in mice [78]. Unlike the other steroids, mineralocorticoids foster the development of fibrosis by upregulating serum- and glucocorticoid-inducible kinase 1 (SGK1), which increases NF-κB, thus promoting fibrosis [84]. Additionally, aldosterone treatment could lead to liver fibrosis in male rats independent of blood pressure via promoting the expression of several fibrosis-associated genes (e.g., TGF-β, α-SMA) [80]. Furthermore, the expression of MR in hepatic stellate cells correlates with inflammation and fibrosis development in choline-deficient and amino-acid-defined diet-induced NASH [79]. Specific MR blockade with eplerenone effectively ameliorated histological steatosis and hepatic fibrosis in a mouse model of NASH. These data provide the basis for therapeutic exploitation of MR blockade for treatment of NASH [79].
Hepatic inflammation can also be affected by vitamin D through downregulation of the expression of Toll-like receptors on Kupffer cells [85]. Vitamin D deficiency causes upregulation of inflammation and oxidative stress genes [81]. Furthermore, restoration of vitamin D could effectively improve TNF-α-modulated immunological abnormalities in a diet-induced steatohepatitis rat model [82]. Moreover, serum vitamin D was significantly decreased in NAFLD patients with advanced liver fibrosis, suggesting that vitamin D might be associated with the progression of liver fibrosis [86]. Further study showed that vitamin D plays an antifibrotic role by suppressing the activation of the HSC-mediated TGF-β signaling pathway, inhibiting the accumulation of profibrotic extracellular matrix proteins [87] and the expression of profibrotic genes including collagen and α-smooth muscle actin (α-SMA) [88][89].
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of Nafld and Nash: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20.
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: An Emerging Menace. J. Hepatol. 2012, 56, 1384–1391.
- Jou, J.; Choi, S.S.; Diehl, A.M. Mechanisms of Disease Progression in Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2008, 28, 370–379.
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology 2011, 140, 124–131.
- Armstrong, M.J.; Adams, L.A.; Canbay, A.; Syn, W.K. Extrahepatic Complications of Nonalcoholic Fatty Liver Disease. Hepatology 2014, 59, 1174–1197.
- Cusi, K. Role of Obesity and Lipotoxicity in the Development of Nonalcoholic Steatohepatitis: Pathophysiology and Clinical Implications. Gastroenterology 2012, 142, 711–725.
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (Nafld). Metabolism 2016, 65, 1038–1048.
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205.
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-Alcoholic Fatty Liver Disease (Nafld) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease. Nutrients 2013, 5, 1544–1560.
- Utzschneider, K.M.; Kahn, S.E. Review: The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761.
- Yang, M.; Zhang, M.; Liu, Q.; Xu, T.; Huang, T.; Yao, D.; Wong, C.W.; Liu, J.; Guan, M. 18beta-Glycyrrhetinic Acid Acts through Hepatocyte Nuclear Factor 4 Alpha to Modulate Lipid and Carbohydrate Metabolism. Pharmacol. Res. 2020, 157, 104840.
- Guan, M.; Qu, L.; Tan, W.; Chen, L.; Wong, C.W. Hepatocyte Nuclear Factor-4 Alpha Regulates Liver Triglyceride Metabolism in Part through Secreted Phospholipase a(2) Gxiib. Hepatology 2011, 53, 458–466.
- Gusdon, A.M.; Song, K.X.; Qu, S. Nonalcoholic Fatty Liver Disease: Pathogenesis and Therapeutics from a Mitochondria-Centric Perspective. Oxid. Med. Cell. Longev. 2014, 2014, 637027.
- Zhang, M.; Yang, M.; Wang, N.; Liu, Q.; Wang, B.; Huang, T.; Tong, Y.; Ming, Y.; Wong, C.W.; Liu, J.; et al. Andrographolide Modulates Hnf4alpha Activity Imparting on Hepatic Metabolism. Mol. Cell. Endocrinol. 2020, 513, 110867.
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of Non-Alcoholic Fatty Liver Disease. QJM 2010, 103, 71–83.
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of Nash. Int. J. Mol. Sci. 2016, 17, 1575.
- Handa, P.; Maliken, B.D.; Nelson, J.E.; Morgan-Stevenson, V.; Messner, D.J.; Dhillon, B.K.; Klintworth, H.M.; Beauchamp, M.; Yeh, M.M.; Elfers, C.T.; et al. Reduced Adiponectin Signaling Due to Weight Gain Results in Nonalcoholic Steatohepatitis through Impaired Mitochondrial Biogenesis. Hepatology 2014, 60, 133–145.
- Younossi, Z.M.; Gramlich, T.; Liu, Y.C.; Matteoni, C.; Petrelli, M.; Goldblum, J.; Rybicki, L.; McCullough, A.J. Nonalcoholic Fatty Liver Disease: Assessment of Variability in Pathologic Interpretations. Mod. Pathol. 1998, 11, 560–565.
- Beste, L.A.; Leipertz, S.L.; Green, P.K.; Dominitz, J.A.; Ross, D.; Ioannou, G.N. Trends in Burden of Cirrhosis and Hepatocellular Carcinoma by Underlying Liver Disease in Us Veterans, 2001-2013. Gastroenterology 2015, 149, 1471–1482.e5.
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic Reticulum Stress and Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Free Radic. Res. 2015, 49, 1405–1418.
- Tarantino, G.; Finelli, C. Pathogenesis of Hepatic Steatosis: The Link between Hypercortisolism and Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2013, 19, 6735–6743.
- Charni-Natan, M.; Aloni-Grinstein, R.; Osher, E.; Rotter, V. Liver and Steroid Hormones-Can a Touch of P53 Make a Difference? Front. Endocrinol. 2019, 10, 374.
- Morris, E.M.; Fletcher, J.A.; Thyfault, J.P.; Rector, R.S. The Role of Angiotensin Ii in Nonalcoholic Steatohepatitis. Mol. Cell. Endocrinol. 2013, 378, 29–40.
- Ali, M.A.; Lacin, S.; Abdel-Wahab, R.; Uemura, M.; Hassan, M.; Rashid, A.; Duda, D.G.; Kaseb, A.O. Nonalcoholic Steatohepatitis-Related Hepatocellular Carcinoma: Is There a Role for the Androgen Receptor Pathway? Onco Targets Ther. 2017, 10, 1403–1412.
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327.
- Chen, K.L.; Madak-Erdogan, Z. Estrogens and Female Liver Health. Steroids 2018, 133, 38–43.
- Hart-Unger, S.; Arao, Y.; Hamilton, K.J.; Lierz, S.L.; Malarkey, D.E.; Hewitt, S.C.; Freemark, M.; Korach, K.S. Hormone Signaling and Fatty Liver in Females: Analysis of Estrogen Receptor Alpha Mutant Mice. Int. J. Obes. 2017, 41, 945–954.
- Zhu, L.; Martinez, M.N.; Emfinger, C.H.; Palmisano, B.T.; Stafford, J.M. Estrogen Signaling Prevents Diet-Induced Hepatic Insulin Resistance in Male Mice with Obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1188–E1197.
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256.
- Yang, M.; Liu, Q.; Huang, T.; Tan, W.; Qu, L.; Chen, T.; Pan, H.; Chen, L.; Liu, J.; Wong, C.W.; et al. Dysfunction of Estrogen-Related Receptor Alpha-Dependent Hepatic Vldl Secretion Contributes to Sex Disparity in Nafld/Nash Development. Theranostics 2020, 10, 10874–10891.
- Della Torre, S. Non-Alcoholic Fatty Liver Disease as a Canonical Example of Metabolic Inflammatory-Based Liver Disease Showing a Sex-Specific Prevalence: Relevance of Estrogen Signaling. Front. Endocrinol. 2020, 11, 572490.
- Chang, H.T.; Pan, H.J.; Lee, C.H. Prevention of Tamoxifen-Related Nonalcoholic Fatty Liver Disease in Breast Cancer Patients. Clin. Breast Cancer 2018, 18, e677–e685.
- Lin, H.Y.; Yu, I.C.; Wang, R.S.; Chen, Y.T.; Liu, N.C.; Altuwaijri, S.; Hsu, C.L.; Ma, W.L.; Jokinen, J.; Sparks, J.D.; et al. Increased Hepatic Steatosis and Insulin Resistance in Mice Lacking Hepatic Androgen Receptor. Hepatology 2008, 47, 1924–1935.
- Lemke, U.; Krones-Herzig, A.; Berriel Diaz, M.; Narvekar, P.; Ziegler, A.; Vegiopoulos, A.; Cato, A.C.; Bohl, S.; Klingmuller, U.; Screaton, R.A.; et al. The Glucocorticoid Receptor Controls Hepatic Dyslipidemia through Hes1. Cell Metab. 2008, 8, 212–223.
- D’Souza, A.M.; Beaudry, J.L.; Szigiato, A.A.; Trumble, S.J.; Snook, L.A.; Bonen, A.; Giacca, A.; Riddell, M.C. Consumption of a High-Fat Diet Rapidly Exacerbates the Development of Fatty Liver Disease That Occurs with Chronically Elevated Glucocorticoids. Am. J. Physiol Gastrointest. Liver Physiol. 2012, 302, G850–G863.
- Zhang, Y.Y.; Li, C.; Yao, G.F.; Du, L.J.; Liu, Y.; Zheng, X.J.; Yan, S.; Sun, J.Y.; Liu, Y.; Liu, M.Z.; et al. Deletion of Macrophage Mineralocorticoid Receptor Protects Hepatic Steatosis and Insulin Resistance through Eralpha/Hgf/Met Pathway. Diabetes 2017, 66, 1535–1547.
- Luo, P.; Dematteo, A.; Wang, Z.; Zhu, L.; Wang, A.; Kim, H.S.; Pozzi, A.; Stafford, J.M.; Luther, J.M. Aldosterone Deficiency Prevents High-Fat-Feeding-Induced Hyperglycaemia and Adipocyte Dysfunction in Mice. Diabetologia 2013, 56, 901–910.
- Marziou, A.; Philouze, C.; Couturier, C.; Astier, J.; Obert, P.; Landrier, J.F.; Riva, C. Vitamin D Supplementation Improves Adipose Tissue Inflammation and Reduces Hepatic Steatosis in Obese C57bl/6j Mice. Nutrients 2020, 12, 342.
- Zhu, C.G.; Liu, Y.X.; Wang, H.; Wang, B.P.; Qu, H.Q.; Wang, B.L.; Zhu, M. Active Form of Vitamin D Ameliorates Non-Alcoholic Fatty Liver Disease by Alleviating Oxidative Stress in a High-Fat Diet Rat Model. Endocr. J. 2017, 64, 663–673.
- Jaruvongvanich, V.; Sanguankeo, A.; Riangwiwat, T.; Upala, S. Testosterone, Sex Hormone-Binding Globulin and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Ann. Hepatol. 2017, 16, 382–394.
- Kim, S.; Kwon, H.; Park, J.H.; Cho, B.; Kim, D.; Oh, S.W.; Lee, C.M.; Choi, H.C. A Low Level of Serum Total Testosterone Is Independently Associated with Nonalcoholic Fatty Liver Disease. BMC Gastroenterol. 2012, 12, 69.
- Eguchi, Y.; Eguchi, T.; Mizuta, T.; Ide, Y.; Yasutake, T.; Iwakiri, R.; Hisatomi, A.; Ozaki, I.; Yamamoto, K.; Kitajima, Y.; et al. Visceral Fat Accumulation and Insulin Resistance Are Important Factors in Nonalcoholic Fatty Liver Disease. J. Gastroenterol. 2006, 41, 462–469.
- Ma, W.L.; Lai, H.C.; Yeh, S.; Cai, X.; Chang, C. Androgen Receptor Roles in Hepatocellular Carcinoma, Fatty Liver, Cirrhosis and Hepatitis. Endocr. Relat. Cancer 2014, 21, R165–R182.
- Wang, J.C.; Gray, N.E.; Kuo, T.; Harris, C.A. Regulation of Triglyceride Metabolism by Glucocorticoid Receptor. Cell Biosci. 2012, 2, 19.
- Sorensen, H.N.; Gautik, K.M.; Bremer, J.; Spydevold, O. Induction of the Three Peroxisomal Beta-Oxidation Enzymes Is Synergistically Regulated by Dexamethasone and Fatty Acids, and Counteracted by Insulin in Morris 7800c1 Hepatoma Cells in Culture. Eur. J. Biochem. 1992, 208, 705–711.
- Andrews, R.C.; Walker, B.R. Glucocorticoids and Insulin Resistance: Old Hormones, New Targets. Clin. Sci. 1999, 96, 513–523.
- Rockall, A.G.; Sohaib, S.A.; Evans, D.; Kaltsas, G.; Isidori, A.M.; Monson, J.P.; Besser, G.M.; Grossman, A.B.; Reznek, R.H. Hepatic Steatosis in Cushing’s Syndrome: A Radiological Assessment Using Computed Tomography. Eur. J. Endocrinol. 2003, 149, 543–548.
- Belden, Z.; Deiuliis, J.A.; Dobre, M.; Rajagopalan, S. The Role of the Mineralocorticoid Receptor in Inflammation: Focus on Kidney and Vasculature. Am. J. Nephrol. 2017, 46, 298–314.
- Barchetta, I.; Carotti, S.; Labbadia, G.; Gentilucci, U.V.; Muda, A.O.; Angelico, F.; Silecchia, G.; Leonetti, F.; Fraioli, A.; Picardi, A.; et al. Liver Vitamin D Receptor, Cyp2r1, and Cyp27a1 Expression: Relationship with Liver Histology and Vitamin D3 Levels in Patients with Nonalcoholic Steatohepatitis or Hepatitis C Virus. Hepatology 2012, 56, 2180–2187.
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D and Metabolic Dysfunction-Associated Fatty Liver Disease (Mafld): An Update. Nutrients 2020, 12, 3302.
- Kim, K.H.; Lee, M.S. Pathogenesis of Nonalcoholic Steatohepatitis and Hormone-Based Therapeutic Approaches. Front. Endocrinol. 2018, 9, 485.
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492.
- Broom, L.J.; Kogut, M.H. Inflammation: Friend or Foe for Animal Production? Poult. Sci. 2018, 97, 510–514.
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435.
- Kamei, Y.; Xu, L.; Heinzel, T.; Torchia, J.; Kurokawa, R.; Gloss, B.; Lin, S.C.; Heyman, R.A.; Rose, D.W.; Glass, C.K.; et al. A Cbp Integrator Complex Mediates Transcriptional Activation and Ap-1 Inhibition by Nuclear Receptors. Cell 1996, 85, 403–414.
- Koyama, Y.; Brenner, D.A. Liver Inflammation and Fibrosis. J. Clin. Investig. 2017, 127, 55–64.
- Parola, M.; Pinzani, M. Liver Fibrosis: Pathophysiology, Pathogenetic Targets and Clinical Issues. Mol. Aspects Med. 2019, 65, 37–55.
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411.
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic Stellate Cells as Key Target in Liver Fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42.
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669.
- Browning, J.D.; Horton, J.D. Molecular Mediators of Hepatic Steatosis and Liver Injury. J. Clin. Investig. 2004, 114, 147–152.
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; Network, N.C.R. Nonalcoholic Fatty Liver Disease (Nafld) Activity Score and the Histopathologic Diagnosis in Nafld: Distinct Clinicopathologic Meanings. Hepatology 2011, 53, 810–820.
- Florentino, G.S.; Cotrim, H.P.; Vilar, C.P.; Florentino, A.V.; Guimaraes, G.M.; Barreto, V.S. Nonalcoholic Fatty Liver Disease in Menopausal Women. Arq. Gastroenterol. 2013, 50, 180–185.
- Suzuki, A.; Abdelmalek, M.F. Nonalcoholic Fatty Liver Disease in Women. Womens Health 2009, 5, 191–203.
- Galmes-Pascual, B.M.; Martinez-Cignoni, M.R.; Moran-Costoya, A.; Bauza-Thorbrugge, M.; Sbert-Roig, M.; Valle, A.; Proenza, A.M.; Llado, I.; Gianotti, M. 17beta-Estradiol Ameliorates Lipotoxicity-Induced Hepatic Mitochondrial Oxidative Stress and Insulin Resistance. Free Radic. Biol. Med. 2020, 150, 148–160.
- Kamada, Y.; Kiso, S.; Yoshida, Y.; Chatani, N.; Kizu, T.; Hamano, M.; Tsubakio, M.; Takemura, T.; Ezaki, H.; Hayashi, N.; et al. Estrogen Deficiency Worsens Steatohepatitis in Mice Fed High-Fat and High-Cholesterol Diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G1031–G1043.
- Xin, G.; Qin, S.; Wang, S.; Wang, X.; Zhang, Y.; Wang, J. Sex Hormone Affects the Severity of Non-Alcoholic Steatohepatitis through the Myd88-Dependent Il-6 Signaling Pathway. Exp. Biol. Med. 2015, 240, 1279–1286.
- Ohashi, T.; Kato, M.; Yamasaki, A.; Kuwano, A.; Suzuki, H.; Kohjima, M.; Ogawa, Y. Effects of High Fructose Intake on Liver Injury Progression in High Fat Diet Induced Fatty Liver Disease in Ovariectomized Female Mice. Food Chem. Toxicol. 2018, 118, 190–197.
- Turola, E.; Petta, S.; Vanni, E.; Milosa, F.; Valenti, L.; Critelli, R.; Miele, L.; Maccio, L.; Calvaruso, V.; Fracanzani, A.L.; et al. Ovarian Senescence Increases Liver Fibrosis in Humans and Zebrafish with Steatosis. Dis. Model. Mech. 2015, 8, 1037–1046.
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Yates, K.; Unalp-Arida, A.; Lavine, J.E.; Clark, J.M.; Diehl, A.M.; et al. A Longer Duration of Estrogen Deficiency Increases Fibrosis Risk among Postmenopausal Women with Nonalcoholic Fatty Liver Disease. Hepatology 2016, 64, 85–91.
- Lu, G.; Shimizu, I.; Cui, X.; Itonaga, M.; Tamaki, K.; Fukuno, H.; Inoue, H.; Honda, H.; Ito, S. Antioxidant and Antiapoptotic Activities of Idoxifene and Estradiol in Hepatic Fibrosis in Rats. Life Sci. 2004, 74, 897–907.
- Ponnusamy, S.; Tran, Q.T.; Thiyagarajan, T.; Miller, D.D.; Bridges, D.; Narayanan, R. An Estrogen Receptor Beta-Selective Agonist Inhibits Non-Alcoholic Steatohepatitis in Preclinical Models by Regulating Bile Acid and Xenobiotic Receptors. Exp. Biol. Med. 2017, 242, 606–616.
- Mohamad, N.V.; Wong, S.K.; Wan Hasan, W.N.; Jolly, J.J.; Nur-Farhana, M.F.; Ima-Nirwana, S.; Chin, K.Y. The Relationship between Circulating Testosterone and Inflammatory Cytokines in Men. Aging Male 2019, 22, 129–140.
- Zhang, H.; Liu, Y.; Wang, L.; Li, Z.; Zhang, H.; Wu, J.; Rahman, N.; Guo, Y.; Li, D.; Li, N.; et al. Differential Effects of Estrogen/Androgen on the Prevention of Nonalcoholic Fatty Liver Disease in the Male Rat. J. Lipid Res. 2013, 54, 345–357.
- Morita, A.; Ishigaki, Y. Gender-Difference in Diabetes Mellitus. Nihon Rinsho 2015, 73, 606–610.
- Lanari, A.; Garegnani, T.C.; Heinrichs, G.; Castresana, M.P. [Hepatic Fibrosis and Progesterone]. Acta Gastroenterol. Latinoam. 1988, 18, 161–171.
- Kim, K.H.; Lee, J.M.; Zhou, Y.; Harpavat, S.; Moore, D.D. Glucocorticoids Have Opposing Effects on Liver Fibrosis in Hepatic Stellate and Immune Cells. Mol. Endocrinol. 2016, 30, 905–916.
- Wada, T.; Miyashita, Y.; Sasaki, M.; Aruga, Y.; Nakamura, Y.; Ishii, Y.; Sasahara, M.; Kanasaki, K.; Kitada, M.; Koya, D.; et al. Eplerenone Ameliorates the Phenotypes of Metabolic Syndrome with Nash in Liver-Specific Srebp-1c Tg Mice Fed High-Fat and High-Fructose Diet. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1415–E1425.
- Pizarro, M.; Solis, N.; Quintero, P.; Barrera, F.; Cabrera, D.; Rojas-de Santiago, P.; Arab, J.P.; Padilla, O.; Roa, J.C.; Moshage, H.; et al. Beneficial Effects of Mineralocorticoid Receptor Blockade in Experimental Non-Alcoholic Steatohepatitis. Liver Int. 2015, 35, 2129–2138.
- Queisser, N.; Happ, K.; Link, S.; Jahn, D.; Zimnol, A.; Geier, A.; Schupp, N. Aldosterone Induces Fibrosis, Oxidative Stress and DNA Damage in Livers of Male Rats Independent of Blood Pressure Changes. Toxicol. Appl. Pharmacol. 2014, 280, 399–407.
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D Deficiency in Obese Rats Exacerbates Nonalcoholic Fatty Liver Disease and Increases Hepatic Resistin and Toll-Like Receptor Activation. Hepatology 2012, 55, 1103–1111.
- Nakano, T.; Cheng, Y.F.; Lai, C.Y.; Hsu, L.W.; Chang, Y.C.; Deng, J.Y.; Huang, Y.Z.; Honda, H.; Chen, K.D.; Wang, C.C.; et al. Impact of Artificial Sunlight Therapy on the Progress of Non-Alcoholic Fatty Liver Disease in Rats. J. Hepatol. 2011, 55, 415–425.
- Nolly, M.B.; Caldiz, C.I.; Yeves, A.M.; Villa-Abrille, M.C.; Morgan, P.E.; Amado Mondaca, N.; Portiansky, E.L.; Chiappe de Cingolani, G.E.; Cingolani, H.E.; Ennis, I.L. The Signaling Pathway for Aldosterone-Induced Mitochondrial Production of Superoxide Anion in the Myocardium. J. Mol. Cell. Cardiol. 2014, 67, 60–68.
- Artunc, F.; Lang, F. Mineralocorticoid and Sgk1-Sensitive Inflammation and Tissue Fibrosis. Nephron Physiol. 2014, 128, 35–39.
- Eliades, M.; Spyrou, E. Vitamin D: A New Player in Non-Alcoholic Fatty Liver Disease? World J. Gastroenterol. 2015, 21, 1718–1727.
- Yang, B.B.; Chen, Y.H.; Zhang, C.; Shi, C.E.; Hu, K.F.; Zhou, J.; Xu, D.X.; Chen, X. Low Vitamin D Status Is Associated with Advanced Liver Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Endocrine 2017, 55, 582–590.
- Breitkopf, K.; Godoy, P.; Ciuclan, L.; Singer, M.V.; Dooley, S. Tgf-Beta/Smad Signaling in the Injured Liver. Z. Gastroenterol. 2006, 44, 57–66.
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D Supplementation and Non-Alcoholic Fatty Liver Disease: Present and Future. Nutrients 2017, 9, 1015.
- Beilfuss, A.; Sowa, J.P.; Sydor, S.; Beste, M.; Bechmann, L.P.; Schlattjan, M.; Syn, W.K.; Wedemeyer, I.; Mathe, Z.; Jochum, C.; et al. Vitamin D Counteracts Fibrogenic Tgf-Beta Signalling in Human Hepatic Stellate Cells Both Receptor-Dependently and Independently. Gut 2015, 64, 791–799.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
3 times
(View History)
Update Date:
12 Oct 2021
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No