 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hui Sun | + 1420 word(s) | 1420 | 2021-05-07 09:32:04 | | | |
| 2 | Lindsay Dong | Meta information modification | 1420 | 2021-06-18 06:37:53 | | |
Video Upload Options
A wide variety of neurogenetic diseases have been discovered owing to the development of next-generation sequencing (NGS) technology. The clinical application of NGS significantly accelerated the discovery of disease-causing genes and promoted the understanding of molecular genetic mechanisms associated with hereditary diseases.
1. Introduction
Next-generation sequencing (NGS) technology has led to great advances in understanding the causes of Mendelian and complex neurological diseases. Owing to the complexity of genetic diseases, the genetic factors contributing to many rare and common neurological diseases remain poorly understood. Selecting the correct genetic test based on cost-effectiveness, coverage area, and sequencing range can improve diagnosis, treatments, and prevention. Whole-exome sequencing and whole-genome sequencing are suitable methods for finding new mutations, and gene panels are suitable for exploring the roles of specific genes in neurogenetic diseases.
2. NGS Tools
2.1. Whole-Exome Sequencing
WES employs NGS platforms, such as Illumina, to sequence the protein-coding regions of the genome. Although initial sequencing and analysis of the human genome revealed that less than 2% of the genome comprises exons, approximately 85% of the DNA variations responsible for highly penetrant genetic diseases lie in this small fraction of the genome [1][2]. Currently, WES is the most commonly used mainstream sequencing method in clinical applications due to its low associated cost and turnaround time, compared with WGS.
2.2. Whole-Genome Sequencing
During WGS approaches, DNA is extracted from cell sources—including peripheral blood leukocytes—and cut into several pieces before being linking with engineered DNA to be sequenced (Figure 1 shows a simplified workflow for NGS). The sequencing results are subjected to sophisticated computerized analysis and careful comparison is made with genomic reference sequences (in related databases) to obtain detailed annotation information [3][4]. WGS screens the entire genome—including coding and noncoding regions, regulatory regions, and SVs leading to copy number variations (CNVs)—facilitating the simultaneous examination of active genes and silent sequences for novel genes, variants, de novo mutations, and loci associated with specific traits [3].
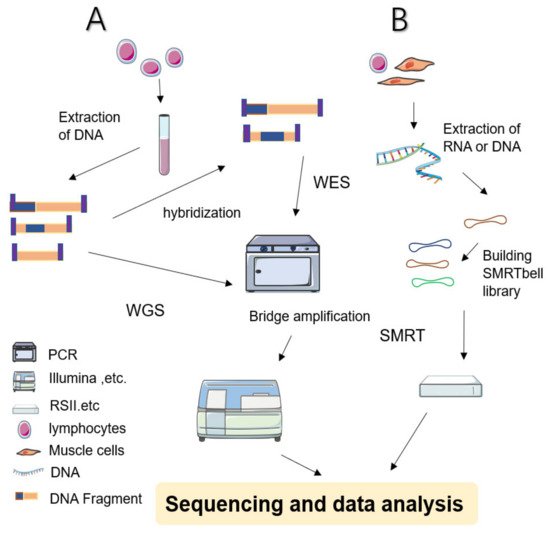
Figure 1. Simplified schematic diagram of NGS and SMRT. (A) WES: Sample preparation begins with extracted and purified DNA which is transposed into fragments. Adapters are tagged before adding motifs. Hybridization is then allowed in the flow cell. After bridge amplification, the Illumina system finally produces the first read. By contrast, WGS does not require hybridized fragments and is ready to be sequenced immediately once the library has been prepared. (B) SMRT: Library preparation begins with a DNA circular structure. When the polymerase encounters a strand of nucleotides containing modifications, the interpulse duration is be delayed. SMRT has two sequencing modes: circular consensus sequencing (CCS) and continuous long read (CLR) sequencing. Because CCS can scroll and copy the same segment along the circular DNA to eliminate errors, its accuracy is higher than 99%. The advantage of CLR is that it can handle longer reads.
2.3. Gene Panels
Gene panels initially capture a set of relevant disease-associated genes, followed by large-scale parallel sequencing [5]. Panels are particularly applicable for studying genetically heterogeneous disorders with well-defined disease-associated genes. These panels can be used to detect approximately 400 related genes for neuromuscular diseases, including congenital myasthenia, congenital myopathy, ataxias, periodic paralysis, motor neuron disorders, spastic paraplegia, Parkinson’s disease, and epilepsy, among others [6][5][7][8][9]. Owing to the low cost and high diagnostic rates, gene panel testing is a common NGS tool used in the field of neurology.
3. Application of Next-Generation Sequencing in Neurogenetic Diseases
3.1. Charcot–Marie–Tooth Disease
CMT encompasses Charcot–Marie–Tooth disease and the related hereditary motor neuropathy (HMN) and hereditary sensory neuropathy disorders, representing the most common group of inherited neuromuscular diseases. CMT is characterized by distal weakness, sensory loss, and a high incidence of foot deformities, including pes cavus [10]. To date, more than 100 genes with pathogenic mutations have been described in relation to CMT. The most common subtype is CMT1A, which accounts for more than 60% of diagnosed cases [11]. Genetic drift contributes to the irregular distribution of different CMT subtypes in each geographical and ethnic population worldwide. In fact, in certain instances, a founder mutation has been shown to be more prevalent. For example, in Slovakia, NDRG1 and HK1 genes, rather than CMT1A, are responsible for the majority of Roma cases in some areas [12]. Hence, the approach taken for diagnosing CMT may differ based on the specific ethnic background of the patient [13].
3.2. Spinocerebellar Ataxias
SCAs generally refer to a group of ataxias with AD inheritance. Additionally, a portion of AR ataxias is designated as SCAR [14]. There are 48 subtypes of SCAs with 36 pathogenetic genes identified to date. More than 100 SCAR genes have also been revealed by NGS [15]. Genetically, SCAs are categorized as either repeat expansion or nonrepeat mutations [14]. Although there is currently no consensus on the optimum order of genetic tests for SCAs, it is recommended to first test for CAG repeat expansions due to their high prevalence. If the result is negative, gene panels and WES can then be considered according to the specific situation [14][15]. A retrospective study published in 2021 estimated the diagnostic yield in 124 SCA patients from 102 families in Italy, with a reported total diagnosis rate of 52%. AD-SCA patients had the highest diagnostic yield (64.5%) among the SCA cases, of which the most frequent subtype was SCA2, followed by SCA1, SCA3, SCA6, and SCA7 [16]. Furthermore, the genetic epidemiology of SCA, like that of any rare disease, is affected by founder effect. For instance, SCA3 is the most common subtype globally, as well as in Western Europe, but has not been identified in Poland. Interestingly, SCA1, caused by ATXN1 gene mutations, is the most prevalent subtype in Poland compared with any other country and may, thus, represent a potential founder effect [17].
Gene panels and WES have served as helpful tools for the diagnosis of AD ataxias (Table 1). SCA6 is caused by CAG-repeat expansion in exon 47 of the CACNA1A gene [18]. Specifically, Saathoff et al. [19] revealed a new pathogenic nonsense variant in CACNA1A (c.2983G>T), which exhibited segregation with the disease for other family members with cerebellar syndrome. The case indicated that this nonsense variant might cause the disease by producing a truncated protein or via nonsense-mediated mRNA decay.
3.3. Epilepsy
Using NGS platforms, several genes encoding voltage-gated ion channels were defined as being associated with epileptic encephalopathies (EE) and developmental and epileptic encephalopathies (DEE) [20][21]. Inuzuka et al. [22] described a patient with an uncommon form of hyperkinetic focal motor seizure in EE carrying a newly discovered variant of KCNT2 which affected the putative pore-forming domain of the protein. A group of diseases characterized by monogenic inheritance and developmental disorders, designated (DEE), is a main beneficiary of NGS [23]. KCNA2 is a DEE-associated gene that encodes the voltage-gated K+ channel KV1.2 [20].
3.4. Multiple Sclerosis
The application of NGS in MS currently focuses on identifying microRNAs (miRNAs), which participate in the pathogenesis of MS through various biological processes influencing immune cells in innate and adaptive immunity [24]. Researchers found that four circulating miRNA exosome sequences were differentially expressed in relapsing-remitting multiple sclerosis (RRMS) patients compared with healthy controls. These results indicated that miRNAs are expected to become a biomarker for predicting and distinguishing MS relapse [25]. Moreover, secondary progressive multiple sclerosis (SPMS) causes modest immune activation compared with RRMS. In fact, an NGS study revealed that miRNA expression declined in the CD4+ T cells of SPMS patients [26].
4. Concluding Remarks and Future Perspectives
NGS is an integral component for delivering precise therapy options to patients. Indeed, NGS may one day replace some of the current methods used for disease diagnosis. An example of this was demonstrated by a population-based study which reported that the highest epilepsy diagnostic yield was obtained via MRI (65%), and, although WES/WGS was performed in only 26/116 cases (22%), the associated diagnostic yield reached 58% [27]. Additionally, the diagnosis of certain conditions associated with neuromyopathy, such as MD, and CMS can avoid the need for muscle biopsy by instead performing genetic testing [28]. Moreover, NGS may become a tool for early intervention or trial treatment, thereby greatly reversing disease trajectory [29][30]. As hereditary diseases account for a significant proportion of morbidity and mortality in infants, rapid whole-genome sequencing can be employed as a primary test for critically ill newborns, thus accelerating the delivery of effective treatments and reducing medical costs [30]. Meanwhile, NGS is gradually realizing accurate symptomatic treatment—perfect examples of which are advances in treatment for Walker–Warburg syndrome and late-onset Pompe disease [31]. This allows doctors to provide accurate advice on treatment options, long-term outcomes, and rehabilitation needs [32]. Selecting the correct genetic test based on cost-effectiveness, coverage area, and optimal test time ensures that clinicians can accurately assess the risk to other family members or future generations and provide genetic guidance.
References
- Winer, L.; Srinivasan, D.; Chun, S.; Lacomis, D.; Jaffa, M.; Fagan, A.; Holtzman, D.M.; Wancewicz, E.; Bennett, C.F.; Bowser, R.; et al. SOD1 in cerebral spinal fluid as a pharmacodynamic marker for antisense oligonucleotide therapy. JAMA Neurol. 2013, 70, 201–207.
- Wang, X.; Shen, X.; Fang, F.; Ding, C.H.; Zhang, H.; Cao, Z.H.; An, D.Y. Phenotype-driven virtual panel is an effective method to analyze WES data of neurological disease. Front. Pharmacol. 2019, 9, 1529.
- Biesecker, L.G. Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: Lessons from the ClinSeq project. Genet. Med. 2012, 14, 393–398.
- Westerink, J.; Visseren, F.L.; Spiering, W. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 2014, 371, 1169.
- Klein, C.J.; Foroud, T.M. Neurology individualized medicine: When to use next-generation sequencing panels. Mayo Clin. Proc. 2017, 92, 292–305.
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38.
- Antoniadi, T.; Buxton, C.; Dennis, G.; Forrester, N.; Smith, D.; Lunt, P.; Burton-Jones, S. Application of targeted multi-gene panel testing for the diagnosis of inherited peripheral neuropathy provides a high diagnostic yield with unexpected phenotype-genotype variability. BMC Med. Genet. 2015, 16, 84.
- Wang, W.; Wang, C.; Dawson, D.B.; Thorland, E.C.; Lundquist, P.A.; Eckloff, B.W.; Wu, Y.; Baheti, S.; Evans, J.M.; Scherer, S.S.; et al. Target-enrichment sequencing and copy number evaluation in inherited polyneuropathy. Neurology 2016, 86, 1762–1771.
- Stehlíková, K.; Skálová, D.; Zídková, J.; Haberlová, J.; Voháňka, S.; Mazanec, R.; Mrázová, L.; Vondráček, P.; Ošlejšková, H.; Zámečník, J.; et al. Muscular dystrophies and myopathies: The spectrum of mutated genes in the Czech Republic. Clin. Genet. 2017, 91, 463–469.
- Reilly, M.M.; Murphy, S.M.; Laurá, M. Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2011, 16, 1–14.
- Rossor, A.M.; Tomaselli, P.J.; Reilly, M.M. Recent advances in the genetic neuropathies. Curr. Opin. Neurol. 2016, 29, 537–548.
- Gabrikova, D.; Mistrik, M.; Bernasovska, J.; Bozikova, A.; Behulova, R.; Tothova, I.; Macekova, S. Founder mutations in NDRG1 and HK1 genes are common causes of inherited neuropathies among Roma/Gypsies in Slovakia. J. Appl. Genet. 2013, 54, 455–460.
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-generation sequencing in Charcot-Marie-Tooth disease: Opportunities and challenges. Nat. Rev. Neurol. 2019, 15, 644–656.
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Primers 2019, 5, 24.
- Manto, M.; Gandini, J.; Feil, K.; Strupp, M. Cerebellar ataxias: An update. Curr. Opin. Neurol. 2020, 33, 150–160.
- Riso, V.; Rossi, S.; Nicoletti, T.F.; Tessa, A.; Travaglini, L.; Zanni, G.; Aiello, C.; Perna, A.; Barghigiani, M.; Pomponi, M.G.; et al. Application of a clinical workflow may lead to increased diagnostic preci-sion in hereditary spastic paraplegias and cerebellar ataxias: A single center experience. Brain Sci. 2021, 11, 246.
- Krysa, W.; Sulek, A.; Rakowicz, M.; Szirkowiec, W.; Zaremba, J. High relative frequency of SCA1 in Poland reflecting a potential founder effect. Neurol. Sci. 2016, 37, 1319–1325.
- Jacobi, H.; du Montcel, S.T.; Bauer, P.; Giunti, P.; Cook, A.; Labrum, R.; Parkinson, M.H.; Durr, A.; Brice, A.; Charles, P.; et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: A longitudinal cohort study. Lancet Neurol. 2015, 14, 1101–1108.
- Saathoff, Y.; Biskup, S.; Funke, C.; Roth, C. New nonsense variant c.2983G>T; p.Glu995* in the CACNA1A gene causes progressive autosomal dominant ataxia. J. Mov. Disord. 2021, 14, 70–74.
- Masnada, S.; Hedrich, U.B.S.; Gardella, E.; Schubert, J.; Kaiwar, C.; Klee, E.W.; Lanpher, B.C.; Gavrilova, R.H.; Synofzik, M.; Bast, T.; et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain 2017, 140, 2337–2354.
- Ambrosino, P.; Soldovieri, M.V.; Bast, T.; Turnpenny, P.D.; Uhrig, S.; Biskup, S.; Döcker, M.; Fleck, T.; Mosca, I.; Manocchio, L.; et al. De novo gain-of-function variants in KCNT2 as a novel cause of developmental and epileptic encephalopathy. Ann. Neurol. 2018, 83, 1198–1204.
- Inuzuka, L.M.; Macedo-Souza, L.I.; Della-Ripa, B.; Monteiro, F.P.; Ramos, L.; Kitajima, J.P.; Garzon, E.; Kok, F. Additional observation of a de novo pathogenic variant in KCNT2 leading to epileptic encephalopathy with clinical features of frontal lobe epilepsy. Brain Dev. 2020, 42, 691–695.
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE commission for classification and terminology. Epilepsia 2017, 58, 512–521.
- Zhang, L.; Wu, H.; Zhao, M.; Chang, C.; Lu, Q. Clinical significance of miRNAs in autoimmunity. J. Autoimmun. 2020, 109, 102438.
- Selmaj, I.; Cichalewska, M.; Namiecinska, M.; Galazka, G.; Horzelski, W.; Selmaj, K.W.; Mycko, M.P. Global exosome transcriptome profiling reveals biomarkers for multiple sclerosis. Ann. Neurol. 2017, 81, 703–717.
- Ogawa, K.; Okuno, T.; Hosomichi, K.; Hosokawa, A.; Hirata, J.; Suzuki, K.; Sakaue, S. Next-generation sequencing identifies contribution of both class I and II HLA genes on susceptibility of multiple sclerosis in Japanese. J. Neuroinflammation 2019, 16, 162.
- Stödberg, T.; Tomson, T.; Barbaro, M.; Stranneheim, H.; Anderlid, B.M.; Carlsson, S.; Åmark, P.; Wedell, A. Epilepsy syndromes, etiologies, and the use of next-generation sequencing in epilepsy presenting in the first 2 years of life: A population-based study. Epilepsia 2020, 61, 2486–2499.
- Gomez-Vargas, A.; Baker, S.K. Molecular diagnosis of myopathies. Rheum. Dis. Clin. North Am. 2011, 37, 269–287.
- Petrikin, J.E.; Willig, L.K.; Smith, L.D.; Kingsmore, S.F. Rapid whole genome sequencing and precision neonatology. Semin. Perinatol. 2015, 39, 623–631.
- Farnaes, L.; Hildreth, A.; Sweeney, N.M.; Clark, M.M.; Chowdhury, S.; Nahas, S.; Cakici, J.A.; Benson, W.; Kaplan, R.H.; Kronick, R.; et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom. Med. 2018, 3, 10.
- Angelini, C.; Savarese, M.; Fanin, M.; Nigro, V. Next generation sequencing detection of late onset pompe disease. Muscle Nerve 2016, 53, 981–983.
- Liu, Z.; Zhu, L.; Roberts, R.; Tong, W. Toward clinical implementation of next-generation sequencing-based genetic testing in rare diseases: Where are we? Trends Genet. 2019, 35, 852–867.

