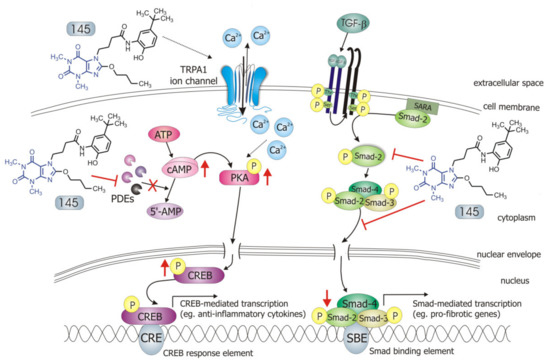
Phosphodiesterases (PDEs) inhibitors are currently a widespread and extensively studied group of anti-inflammatory and anti-fibrotic compounds which may find use in the treatment of numerous lung diseases, including asthma and chronic obstructive pulmonary disease. Several PDE inhibitors are currently in clinical development, and some of them, e.g. roflumilast, are already recommended for clinical use. Due to numerous reports indicating that elevated intracellular adenosine 3’,5’-cyclic monophosphate (cAMP) levels may contribute to alleviation of inflammation and airway fibrosis, new and effective PDE inhibitors are constantly being sought. Recently, a group of 7,8-disubstituted purine-2,6-dione derivatives, representing novel and prominent pan-PDE inhibitors has been synthesized. Some of them were reported to modulate transient receptor potential ankyrin 1 (TRPA1) ion channels as well. Compound 145 (4-(8-butoxy-1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N-(5-(tert-butyl)-2-hydroxyphenyl)butanamide) – a pan-PDE inhibitor and a weak TRPA1 modulator inhibited fibroblast to myofibroblasts transition (FMT) as well as MRC-5 human lung fibroblasts proliferation, migration, and contraction. The effect of this compound appeared to depend mainly on its strong PDE inhibitory properties, and not on its effects on TRPA1 modulation. The strong anti-remodelling effects of 145 required activation of the cAMP/protein kinase A/CREB pathway leading to inhibition of transforming growth factor type β1 (TGF-β1) and Smad-dependent signalling in MRC-5 cells.
Tissue remodeling is a widespread pathological process in which a number of structural changes occur in a tissue or organ that impairs its normal physiological functions [
24,
25,
26]. In the lungs or bronchi, remodeling in asthma, chronic obstructive pulmonary disease (COPD) and lung fibrosis is often associated with the imbalance in extracellular matrix (ECM) protein turnover, disturbances that occur in the structure and functions of the epithelial layer, smooth muscle cell hyperplasia and hypertrophy, and increased levels of tissue myofibroblasts, predominantly as a result of fibroblast to myofibroblast transition [
27,
28]. Therefore, in the search for new anti-fibrotic drugs, particular attention should be paid to pleiotropic compounds that affect various cellular processes associated with remodeling. We have recently shown novel PDE inhibitors from the group of 7,8-disubstituted purine-2,6-dione derivatives as potent modulators of airway smooth muscle cells (ASMCs) remodeling [
19]. Here we demonstrated for the first time that a strong pan-PDE inhibitor from this group, compound 145 can also exert anti-fibrotic effects in TGF-β
1-induced human lung fibroblasts via limiting FMT.
A number of studies have earlier demonstrated FMT-limiting properties of cAMP elevating agents, such as PDE inhibitors or activators of soluble adenylyl cyclase [
29,
30,
31]. The potential benefits of using theophylline as an anti-inflammatory drug in the treatment of asthma have long been known, but it was only a few years ago when Yano et al. reported the anti-FMT effects of this substance in the lung [
32]. In turn, we demonstrated that other methylxanthines, i.e., pentoxifylline or lisofylline, can also reduce FMT in bronchial fibroblasts isolated from asthmatics [
33]. Other studies revealed that selective inhibitors, particularly those inhibiting PDE4, such as rolipram or roflumilast, also show anti-FMT effects [
31,
34,
35].
Our recently designed and synthesized 7,8-disubstituted purine-2,6-dione derivatives fill very well the gap between inhibitors highly selective for a single isoenzyme, and non-selective and weak non-specific inhibitors. Both our previous [
19] and current research show that 7,8-disubstituted purine-2,6-dione derivatives represent a group of pan-PDE inhibitors with high inhibitory potency against individual PDE isoenzymes. They can effectively and strongly inhibit many different PDEs, including those relevant to the pathogenesis of respiratory diseases [
1,
3]. Recently, we confirmed 145 favorable binding modes in the active site of PDE4B and PDE7A as well as to the TRPA1 model [
20]. Moreover, molecular docking studies of close analogs of compounds 832 and 869, which recognized crucial interactions in PDE4B/PDE7A and TRPA1 sites, respectively, were previously described [
20,
21]. Here, it was demonstrated for the first time that these potent, pan-PDE inhibitors exert a strong limiting effect on TGF-β
1-induced FMT in lung fibroblasts. Selected 7,8-disubstituted purine-2,6-dione derivatives, especially 145 inhibited the expression of genes and proteins characteristic for both myofibroblasts and extracellular matrix proteins secreted by these cells. We showed that some compounds belonging to 7,8-disubstituted purine-2,6-dione derivatives can also reduce proliferation, migration, and contraction of pulmonary fibroblasts. We also displayed that the observed anti-fibrotic effects of pan-PDE inhibitor, 145, are the result of its impact on the canonical TGF-β signaling pathway. This compound, decreased Smad-2 phosphorylation and subsequent translocation to the nucleus, thereby reducing TGF-β
1-induced and Smad-mediated transcription of profibrotic genes in lung fibroblasts ().
Figure 8. Compound 145 interaction with TGF-β1-induced MRC-5 cells and possible targets related to its anti-fibrotic effects. Abbreviations: ATP—adenosine triphosphate; 5′-AMP—5′-adenosine monophosphate; CRE—CREB response element; SBE—Smad binding element; SARA—Smad anchor for receptor activation; PKA - protein kinase A
The Smad-dependent TGF-β signaling pathway has already been identified as a potential therapeutic target for preventing fibrosis in the lung and other organs [
36,
37]. This has been confirmed by our current and previous data, in which we have also shown that in asthmatic fibroblasts, pentoxifylline and lisofylline can limit TGF-β signaling [
33]. Further evidence is provided by Fehrholz et al. who demonstrated in lung epithelial cells that caffeine and rolipram might also affect Smad-dependent TGF-β signaling [
38]. Similar effects have been described for the new drugs used in idiopathic pulmonary fibrosis (IPF) therapy, pirfenidone, and nintedanib [
39]. Although the exact mechanism of action of these anti-inflammatory, antioxidant and anti-remodeling drugs is yet unknown, it is has been found that they can affect, among others, TGF-β/Smad-dependent signaling [
39,
40].
Some studies revealed that increased intracellular cAMP levels could directly affect the canonical TGF-β and Smad-dependent signaling pathway [
41,
42,
43,
44,
45]. After an increased CREB phosphorylation (e.g., in response to the elevated intracellular cAMP levels), a CREB-binding protein (CBP) effectively binds p-CREB resulting in enhancement of its transcriptional activity [
46]. On the other hand, it is known that CBP can directly interact with the Smad protein complex and operate as a Smad transcriptional co-activator [
47]. As such, competition between cAMP-dependent CREB/CBP binding and TGF-β-dependent Smad/CBP binding for the common co-activator CBP [
43,
45] is proposed as the mechanistic explanation for the functional inhibition of Smad-dependent signaling. Compound 145, as a strong PDE inhibitor, elevated cAMP levels in lung fibroblasts, activated PKA and, as a consequence, led to increased CREB phosphorylation, which in turn inhibited Smad signaling (). The presence of 145 caused the most prominent activation of the cAMP/PKA/CREB pathway in TGF-β
1-induced MRC-5 cells. At the same time, this compound had the best inhibitory effect on TGF-β
1/Smad-2 signaling. While a strong activation of cAMP-dependent CREB phosphorylation is not surprising given that 145 elevates intracellular cAMP, its inhibitory effect on Smad-2 phosphorylation indicates new and desirable properties of this compound (). Since other Smad proteins can also regulate TGF-β signaling, the effect of 145 on Smad-3/4 or Smad-1 should also be tested in future studies. The increased efficacy of compound 145 may be the result of the interaction with two independent signaling pathways, both of which promote anti-fibrotic effects. It is known that the reduction of TGF-β-induced and Smad-mediated transcription of profibrotic genes is one of the main goals of anti-fibrotic therapy [
36,
37] and out of the studied compounds, 145 best meets this requirement. Of note, is that there are also other reports that associate CREB phosphorylation and activation of CREB-mediated transcription with anti-inflammatory properties, e.g., increased IL-10 synthesis, inhibition of NF-κB signaling, Th17 cell differentiation, and survival of T
reg cells [
48,
49]. Therefore, it is likely that the high CREB phosphorylation induced by 145, compared to other tested compounds, may explain the prominent anti-fibrotic effects of this compound in TGF-β
1-induced MRC-5.
Previous studies [
20] made us aware that 145 may also interact with TRPA1 channels. In light of the reports indicating that TRPA1 channels may be a potential molecular target for lung diseases [
18,
52,
53], and since caffeine can suppress TRPA1 activity [
54], we examined whether 832, 869, and 145 affect the function of this channel. Our results do not support a very strong agonistic/antagonistic activity of 145 towards TRPA1, but functional analysis revealed that this compound can modify calcium influx in pulmonary fibroblasts in a way a weak agonist would. To assess the involvement of TRPA1 in the observed activity of compound 145, we performed a series of experiments in which we pharmacologically blocked or activated this ion channel and then compared the effects in the presence and absence of 145. The presence of 145 neither affected MRC-5 proliferation or cAMP levels, which indicates that the TRPA1 component may not play an important role in 145-mediated anti-fibrotic effects. We cannot exclude that 145 exerts such a strong inhibitory effect on PDE that the maximum anti-fibrotic potential is already achieved, and thus, any further modulation of TRPA1 activity may be negligible for the final outcome. The slight but noticeable effect of the TRPA1 antagonists—HC-030031 and 869, on some tested TGF-β
1-induced MRC-5 myofibroblast features associated with remodeling may support this notion. This is also in line with several clinical trials in which TRPA1 antagonists are tested to treat respiratory diseases [
53]. On the other hand, Kurahara et al. demonstrated that pirfenidone might exert anti-inflammatory and anti-fibrotic effects upon TRPA1 activation [
55]. We have not seen similar effects in our study; in fact, TRPA1 activation via ASP 7663 did not result in significant changes in the phenotype of TGF-β
1-induced MRC-5 cells. Considering the above reports, and since large and sustained intracellular Ca
2+ elevations are often noxious for cells and may trigger cell death [
56], the fact that compound 145 does not excessively activate Ca
2+ influx via TRPA1 might be desirable from the therapeutic perspective. Collectively, it could be argued that anti-fibrotic properties of compound 145 are accomplished by a strong PDE inhibitor activity, while the TRPA1 channel modulation is minor and may only slightly contribute to 145 activity.
Our previous research demonstrated that compounds from the group of 7,8-disubstituted purine-2,6-dione derivatives are able to limit ASMC remodeling response, and here we confirm 145 preferential activity in reducing lung fibroblast features associated with remodeling. Although both the previous and current studies provide solid evidence of the anti-fibrotic effect of compound 145, the studies so far have been performed in vitro in the presence of one growth factor. Therefore, the activity of 145, a new pan-PDE inhibitor, should be verified in vivo in a model of chronic lung diseases and compared with known anti-inflammatory drugs or PDE inhibitors already used clinically. In the in vivo model, it would be possible to know the 145 pleiotropic effects. Given the complexity of bronchial and lung remodeling, the search for compounds with a wide spectrum of activity is extremely valuable. Considering the data obtained with ASMCs and presented here lung fibroblasts, it can be concluded that the group of 7,8-disubstituted purine-2,6-dione derivatives offers promising drug candidates targeting chronic lung disease therapy.
 +1 credit
+1 credit