Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gvozden Rosic | + 3315 word(s) | 3315 | 2021-06-09 08:07:34 | | | |
| 2 | Vivi Li | + 7 word(s) | 3322 | 2021-06-17 03:46:47 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rosic, G. Galectin-3 in Neurodevelopment. Encyclopedia. Available online: https://encyclopedia.pub/entry/10896 (accessed on 28 June 2026).
Rosic G. Galectin-3 in Neurodevelopment. Encyclopedia. Available at: https://encyclopedia.pub/entry/10896. Accessed June 28, 2026.
Rosic, Gvozden. "Galectin-3 in Neurodevelopment" Encyclopedia, https://encyclopedia.pub/entry/10896 (accessed June 28, 2026).
Rosic, G. (2021, June 16). Galectin-3 in Neurodevelopment. In Encyclopedia. https://encyclopedia.pub/entry/10896
Rosic, Gvozden. "Galectin-3 in Neurodevelopment." Encyclopedia. Web. 16 June, 2021.
Copy Citation
There is a plethora of evidence to suggest that Galectin-3 plays an important role in normal functions of mammalian cells, as well as in different pathogenic conditions.
Galectin-3
neurodevelopment
neuroinflammation
behavior
1. Structure and Function of Galectin-3
Galectin-3, which is a sole representative of chimera type, contains one CRD which is linked to the N-terminal domain that allows oligomerization resulting in formation of pentamers. Specifically, upon interaction of Galectin-3 monomers with glycoproteins or glycolipids they interconnect to form pentameric complex by their N-terminal domains (Figure 1).
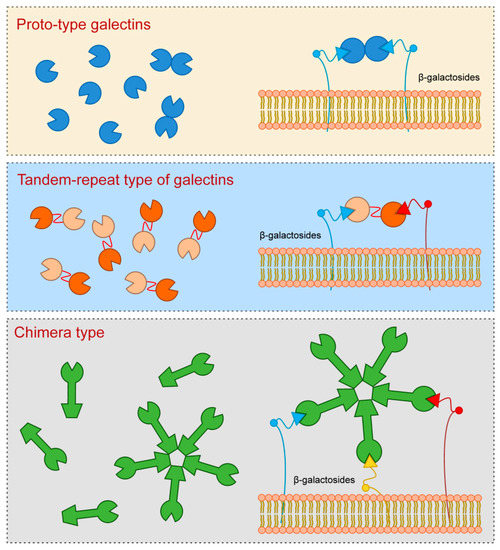
Figure 1. Galectins classification (according to their structures).
Galectin-3 is a protein with approximate molecular weight of 31 kDa, first recognized in murine immune cells, and thereafter found in a variety of normal and tumor cells [1][2][3]. The structure of Galectin-3 is unique among all vertebrate galectins, consisting of two structurally specific domains: N-terminal domain and C-terminal CRD [4]. N-terminal domain usually contains up to 150 amino acid residues, depending on the species, built up of nine repetitive sequences rich in proline, glycine, tyrosine and glutamine [1][2][5]. N-terminal domain carries sites for phosphorylation and other determinants involved in regulation of galectin secretion [6][7][8]. C-terminal CRD, consisting of about 135 amino acid residues, and determines the molecule as a galectin family member. CRD is connected to a collagen-like sequence, assembled of proline, glycine and tyrosine tandem repeats [9].
In adult humans Galectin-3 is present in many different types of cells and tissues. During the first trimester of human embryo development, Galectin-3 is mainly expressed in epithelia, such as lining epithelium of the respiratory system and digestive tract, urothelium, skin, as well as in myocardium, liver and chondrocytes [10]. In adults Galectin-3 is also found in various immune cells, except resting lymphocytes [1][4][11]. Apart from physiological functions in a variety of biological processes such as cell adhesion, cell activation, cell growth and differentiation, cell cycle, and apoptosis, Galectin-3 also has pivotal roles in cell to cell interactions [4][12][13][14].
Galectin-3 synthesis takes place on free ribosomes in the cytoplasm. It can be found in nucleus, on the cell surface and in the extracellular space [15][16]. Given that Galectin-3, as all galectins, lacks a signal sequence that would guide its translocation to endoplasmic reticulum and further enable classical secretory pathway, the secretion of Galectin-3 takes place in a non-classical fashion [16]. Depending on the cell type, Galectin-3 is found in exosomes or microvescicles [17][18][19]. It has recently been shown that endosomal sorting complex required for transport (ESCRT) machinery plays a crucial role in Galectin-3 transport to the extracellular matrix. Specifically, the ESCRT-I component Tsg101 binds to highly conserved P(S/T)AP motif located to N-terminal domain of Galectin-3 allowing the packing of Galectin-3 into endosomes [20]. Biological effects of Galectin-3 are largely determined by its cellular localization, specific tissue, or specific pathological condition. The most exhaustively studied role of Galectin-3 concerns the regulation of inflammatory processes. Pathogenesis of xenobiotic induced Primary Biliary Cholangitis (PBC) indicated protective role of Galectin-3, since PBC in Galectin-3 knock-out (KO) C57Bl/6 mice showed enhanced liver infiltration with CD8+ T lymphocytes followed by augmented bile duct damage, liver fibrosis, serological level of PDC-E2 (E2 component of the pyruvate dehydrogenase complex—common auto-antigen in PBC) specific IgA and increased AST/ALT ratio [21]. Conversely, recent study by Arsenijevic and colleagues pointed out detrimental role of Galectin-3 in PBC with infectious etiology [22]. In Novosphingobium aromaticivorans (N. aromaticivorans) induced PBC, Galectin-3 deletion had anti-inflammatory role, due to the decreased activation of dendritic cells and macrophages in Galectin-3 KO C57Bl/6 mice. Furthermore, we showed that in mouse experimental model of autoimmune myocarditis Galectin-3 had a protective role on disease development [23]. In myosin peptide-induced experimental autoimmune myocarditis (EAM) on C57Bl/6 mice, Galectin-3 KO mice developed more severe myocardial inflammation and more conspicuous hypertrophy, due to the accumulation of T helper type 2 (Th2) cells and expansion of type 2 inflammation in the hearts of otherwise predominantly Th1 C57Bl/6 mice.
2. Galectin-3 and Neurodevelopment
Galectin-3 plays an important role in physiological functions of the nervous system, but it is also implicated in variety of neurological disorders. In normal adult rats Galectin-3 is constitutively expressed both in glial cells and neuronal tissues in different brain regions [24]. There is co-expression of Galectin-3 with specific antigens for different cells in rat brain: NeuN (neuronal nuclear antigen), GFAP (glial fibrillary acidic protein), Iba1 (ionized calcium-binding adapter molecule 1). Immunoreactivity for galectin-3 was shown in several regions of telencephalon (some parts of cerebral cortex with variations in the laminar distribution and regions of amygdala, basal ganglia and septum), diencephalon (thalamus and hypothalamus), brain stem and cerebellum (mesencephalon, rhombencephalon, myelencephalon and cerebellum) [24]. Comte and coworkers [25] indicated the role of Galectin-3 in normal neurodevelopment of mouse brain. Galectin-3 affects migration of neuroblasts from subventricular zone (SVZ) through rostral migratory stream (RMS) towards the olfactory bulb (OB) [25]. In Galectin-3 KO mice migration of neuroblasts was disrupted due to the decreased speed and straightness of migration. One of possible mechanisms implies the increased phosphorylation of epidermal growth factor receptor (EGFR) in the absence of Galectin-3 and its increased activation. Keratinocytes with depleted Galectin-3 have impaired migration and decreased surface EGFR expression [26][27].
Galectin-3 has an important role in oligodendrocyte differentiation and maintenance of myelin integrity and function [28]. In vitro study pointed out that oligodendrocytes express Galectin-3 at various stages of differentiation. In vivo experiments also indicated that Galectin-3 mediates oligodendrocyte differentiation by microglial cells and astrocytes [28]. Furthermore, electron microscopic analysis of myelin showed disturbances in myelination process in Galectin-3 KO mice compared to WT. Thomas and Pasquini showed that Galectin-3 mediated glial crosstalk drives oligodendrocytes differentiation [29].
Asphyxia, followed by hypoxic and ischemic brain damage remains one of the most common causes of neurological disorders in infants [30]. Brain damage induced by hypoxia and ischemia results in activation of the immune system and consequent increase in production of pro-inflammatory cytokines and reactive oxygen species [31][32][33]. Microglial cells have the crucial role in this neuroinflammation. They also produce Galectin-3 which has pro-inflammatory effects in both hypoxia and ischemia [34][35].
In a study on newborn mice which were subjected to neonatal hypoxia/ischemia, Doverhag and coworkers [36] showed the increased expression of Galectin-3 RNA 8 h, 24 h and 72 h after the injury and Galectin-3 colocalized with Iba-1 in activated microglia close to the injury. Furthermore, Galectin-3-deficient mice were protected compared to their WT littermates, given that neuronal tissue volume loss and regional injury of hippocampus and striatum were significantly reduced in Galectin-3-deficient mice. While there was no statistically significant difference in microglia accumulation between Galectin-3-deficient mice and the WT, there were significantly higher levels of total matrix metalloproteinase 9 (MMP-9) protein levels in WTs suggesting the possibility of modulation of microglia phenotype by Galectin-3 and mechanism of injury attenuation in Galectin-3-deficient mice [36]. Study on C57Bl/6 NADPH oxidase KO mice implicated that hypoxic brain injury increased Galectin-3 levels in NADPH KO mice in comparison to the WT, as well as in injured hemisphere compared to the uninjured hemisphere in both the KO and WT mice [37].
Pesheva and collaborators [38] argue that expression of Galectin-3 in neurons depends on the presence of the nerve growth factor (NGF). The authors used neonatal dorsal root ganglion (DRG) neurons to test the effects of NGF, brain-derived neurotrophic factor (BDNF) and neurotrophin-3 on Galectin-3 expression patterns and type of cells which express Galectin-3. They showed that NGF stimulated DRG neurons and macrophage-like cells increased expression of Galectin-3, suggesting a role in promotion of neurite outgrowth and adhesion of neural cells [38][39]. The authors also proposed that molecular mechanism included the activation of TrkA (Tropomyosin receptor kinase A) receptors, while a later study showed that regulation of Galectin-3 expression was mediated through Ras/MAPK-related signaling pathways [38][40]. On the other hand, the same group of authors showed that staurosporine (a protein kinase inhibitor) induced Galectin-3 expression after 1–5 days in culture of PC12 cells which was not affected by Ras/MAPK pathway inhibitors, suggesting also Ras/MAPK-independent mechanism for regulation of Galectin-3 expression [40].
Umekawa and coauthors [41] compared inflammatory responses to brain injury due to hypoxia/ischemia in immature and adult hippocampus, focusing on the differences between resident microglia and macrophages from circulation. Based on the obtained results the authors concluded that in the immature brain resident microglia activated earlier and caused a more pronounced inflammatory response compared to the infiltrating blood-derived macrophages. Furthermore, Galectin-3 expression was more pronounced in immature brains which can be brought into connection with more prominent inflammatory response in newborn animals [41]. Conversely, a study by Chip and coauthors on the role of Galectin-3 in focal stroke caused by transient occlusion of middle cerebral artery resulted in up-regulation of Galectin-3 in neonatal mice and rats [42]. In Galectin-3—deficient mice more severe loss of tissue occurred compared to the wild type [42]. Furthermore, in Galectin-3 deficient mice some of cytokines and chemokines were changed 72 h after the induction of brain damage, where the levels of interleukin 6 (IL-6) and Granulocyte Colony-Stimulating Factor (G-CSF) decreased, and Macrophage Inflammatory Protein 1α (MIP-1α) and MIP-1β increased.
Novel inflammation-independent role of Galectin-3 is shown in regulation of astrogenesis by alteration of bone morphogenetic protein (BMP) signaling [43]. The authors focused on postnatal lateral subventricular zone, given that periventricular regions of the brain are substantially sensitive to hypoxic ischemia, and applied electroporation to increase or decrease Galectin-3 expression in vivo, and nucleofection in vitro [43]. Subventricular zone is the major source of glial cells during postnatal forebrain development, and Galectin-3 deficiency reduces gliogenesis in postnatal period, while Galectin-3 increase has an opposite effect [43]. Furthermore, Galectin-3 binds to bone morphogenetic protein receptor one alpha (BMPR1α), activates BMP signaling and thus regulates basal gliogenesis [43].
3. Galectin-3 and Neuroinflammation
Galectin-3 KO mice developed by Hsu and colleagues on C57Bl/6 background are widely used for the evaluation of Galectin-3 role in inflammatory response [44]. They do not appear to have a distinct phenotype compared to the WTs. Behavioral characteristics are described by Stajic and coauthors [45] and discussed further in chapter 9.
There has recently been increasing evidence that Galectin-3 plays a role in neuroinflammation and neurodegeneration [46]. Experimental autoimmune encephalomyelitis (EAE) reflects pathological changes in multiple sclerosis in humans, providing a widely accepted model for this disease [47]. We observed that the deletion of Galectin-3 gene attenuates EAE in C57Bl/6 mice [48]. This was attributed to modulation of antigen-presenting cells and subsequent attenuation of inflammatory response in CNS. Following immunization of WT and Galectin-3 with myelin oligodendrocyte glycoprotein peptide (MOG35–55) the severity of diseases was significantly lower in Galectin-3-deficient mice. In addition, production of pro-inflammatory cytokines, IL-17 and IFN-γ, in these mice was reduced, while dendritic cells (DC) produced IL-10 and exhibited Th2 polarization. Galectin-3 is also involved in Interleukin 4 (IL-4) mediated macrophage alternative polarization, thus its effect in EAE may be attributed to its effect on activation and proliferation of microglia [44][48]. Microglia represent immune competent cells in the brain which appear to play the important roles both in diverse homeostatic mechanisms in the nervous tissues and in various pathological conditions which affect brain homeostasis [49]. It has been established that activation of microglia can be a double-edged sword, whereby their profiling depends on many different factors. Reichert and Rotshenker [50] also indicated the role of Galectin-3 in the pathogenesis of EAE. First, they showed increased expression of Galectin-3 in macrophages and microglia in the CNS of mice with EAE. On the other hand, using Copolymer 1 as immunomodulator, they suppressed EAE. This was thought to be due to decreased activation of microglia and macrophages, given that Copolymer 1 induces antigen-specific Th2 response and increased secretion of IL-10, which in turn decreases production of pro-inflammatory cytokines and Galectin-3. It has also been postulated that Galectin-3 may be an important activator of phagocytosis of modified myelin, a necessary stage during its recovery within Wallerian degeneration. There is an increased expression of Galectin-3 in microglia that phagocyte myelin, unlike the microglia that do not phagocyte myelin [51][52][53]. Wallerian degeneration following injury of sciatic nerve in Galectin-3-deficient mice was associated with vigorous increase in inflammatory cytokines, IL-1β and TNF-α, and up-regulation of toll-like receptors (TLR) 2 and 4 [54]. The C57Bl/6 mouse model of focal cortical EAE, which were immunized with MOG and received intracerebral solution of tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) developed large lesions with a high number of Galectin-3-positive inflammatory cells [55]. These cells were classified into two main categories—Galectin-3—positive cells with projections, microglia-derived macrophages, and Galectin-3—positive cells without projections, macrophages derived monocytes. Recent data on cell and stage-specific expression of Galectin-3, in mouse model of EAE induced by pathogenic T-cell transfer, showed increased expression of Galectin-3 in microglia with magnified phagocytic activity in spinal gray matter during progressive disease [56]. Furthermore, the expression of Galectin-3 increased in microglia and macrophages in spinal white matter and pia mater during disease progression, while in nerve roots subpopulation of Schwann cells were Galectin-3—positive. During recovery phase, Galectin-3—expressing cells disappeared from parenchyma, and were confined to the pia mater and ventral nerve roots. These results suggest the possibility of neuroprotective role of Galectin-3 in brain pathology, thus Galectin-3 may have both pro and anti-inflammatory effects in the CNS. Its role appears to depend on the type of cells Galectin-3 expresses and its cellular localization. In addition, the effects of Galectin-3 in type 1 and 2 diabetes can be compared when expressed on immune cells and overexpressed in target cells. We have shown previously that Galectin-3 deletion attenuates type 1 diabetes due to its lack in immune effectors cells [57]. However, in type 2 diabetes intracellular genetic overexpression of Galectin-3 (knock in mice) protects pancreatic β-cells from inflammatory attack [58].
Galectin-3 plays a role in the pathogenesis of viral infections of the CNS. Junin virus-induced encephalitis was induced in C57Bl/6 mice by intracerebral inoculation and it was shown that activated microglial cells and astrocytes express Galectin-3 [59]. In Theiler’s Murine Encephalomyelitis Virus (TMEV) infection Galectin-3 expression increased in the cerebral cortex of in C57Bl/6 and SJL/J mice [60]. Furthermore, Galectin-3 deletion in C57Bl/6 mice reduced the number of activated immune cells after TMEV infection and diminished inflammatory response followed by a partial restoration of SVZ proliferation and increase of SVZ progenitor cells.
4. Galectin-3 in the CNS Injury
Galectin-3 appears to be one of the crucial initiators of microglia activation and proliferation following ischemic brain injury, but role of activation of microglial cells upon brain ischemia remains questionable, whether it could be advantageous or injurious [61]. Following experimental stoke in normal C57Bl/6 mice there was strong increase in Galectin-3 expression in microglia surrounding ischemic lesion, while in quiescent microglial cells Galectin-3 immunoreactivity was depressed [34]. In Galectin-3 KO C57Bl/6 mice depletion of Galectin-3 led to inadequate activation and decreased proliferation of microglia, resulting in decrease of microglial cells in ischemic conditions. This disruption in regulation of microglia activity consequently induced increase in infarct size and number of apoptotic neurons. In proliferating microglia there is co-expression of Galectin-3 and Insulin-like growth factor 1 (IGF-1), while in Galectin-3 KO C57Bl/6 mice there increased protein level of IGF-1 after stroke. Specifically, microglia of Galectin-3 depleted mice were not responsive to IGF-1 and it was implicated that Galectin-3 interacted with IGF-1 receptors thus enabling their crosslink at the membrane surface, delay of their removal by endocytosis, and consequently prolonged signaling [26][34]. It has also been shown that Galectin-3 induced proliferation of endothelial cells and neural progenitors upon ischemic brain injury caused by transient middle cerebral artery occlusion (MCAO) in rats, while inhibition of Galectin-3 with anti-Gal-3 antibody had opposite effects [62]. These results suggest a possible role of microglial Galectin-3 in nerve tissue remodeling through angiogenesis and neurogenesis. Recently it was indicated that intracerebroventricular injection of recombinant Galectin-3 during acute phase of stoke, in model of brain ischemia caused by MCAO, induced alternative activation of microglia, increased secretion of IL-4, and decreased production of pro-inflammatory cytokines (TNF-α, IL-1β, INF-γ, IL-6 and IL-17) [63]. Measuring stroke area four days after induction of ischemia and 72 h after the application of recombinant Galectin-3 showed significant reduction in the size of ischemic lesions in mice receiving recombinant Galectin-3 compared to control.
In a study assessing delayed neuronal death induced by transient ischemia in the hippocampal CA1 region, the authors confirmed there was increased expression of Galectin-3 in microglia with the peak which was achieved 96 h following reperfusion [64]. On the other hand, intra-ischemic hypothermia significantly averted delayed neuronal death as well as expression of Galectin-3 in microglial cells.
Furthermore, investigating the connection between stoke and enteric neuropathy, it was shown that sera from C57BL/6 mice subjected to permanent MCAO induced loss of myenteric neurons in vitro, contrary to sera isolated from Galectin-3—deficient mice with induced stoke [65]. Conversely, myenteric neurons obtained from mice deficient in toll-like receptor 4 (TLR4) were unaffected, and also the application of antagonists of transforming growth factor β-activated kinase 1 (TAK1) or AMP activated kinase (AMPK) prevented loss of myenteric neurons in vitro. Combining the results of this study it can be assumed that the release of Galectin-3 following stroke may induce enteric neuronal cell death via TLR4 activation involving TAK1 and AMPK.
Previous studies focused on relationship between Galectin-3 and TLR family in CNS, in particular TLR2 and TLR4. Galectin-3 possesses great affinity for β-galactoside, and considering that TLR4 structurally contains β-galactosides, Galectin-3 binding to TLR4 occurs [66]. Such binding can induce changes in TLR4 appearance, for instance dimerization or internalization. Inflammatory stimulation in brain, such as intranigral injection of lipopolysaccharide (LPS), triggers endogenous production of Galectin-3, which binds to microglial TLR4 and causes their activation [67] (Figure 2). On the other hand, intranigral LPS injection and consequent neuroinflammation in Galectin-3 KO mice were characterized by reduced expression of pro-inflammatory markers, IL-1β and IL-6, and decreased inflammatory response with neuroprotective effect. Effects of in vitro exposition of microglial cells to soluble Galectin-3 were increase of expression of inducible nitric oxide synthase (iNOS) and stimulation of pro-inflammatory M1 phenotype combined with decrease of anti-inflammatory markers. Expression of TLR4 and Galectin-3 were also increased in burn-induced peripheral neuroinflammation [68]. TLR4 appears to be universal receptor for Galectin-3 in inflammation in different tissues [69]. Galectin-3 seems to be an important mediator in injury-induced innate immune response/TLR2 signaling [34]. In microglial cells of Galectin-3 KO mice there was no up-regulation of TLR2 due to stimulation with glutamate, contrary to WT cells. It has also been shown that LPS induction of Galectin-3 release from microglia enhances microglial phagocytosis [70]. Specifically, secreted Galectin-3 opsonizes cells for phagocytosis via interaction with a phagocytic receptor, Mer tyrosine kinase (MerTK), and this process appears to be necessary for phagocytosis of cells by activated microglia [71].
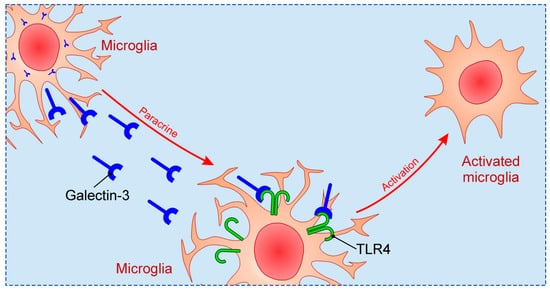
Figure 2. The mechanism of microglial activation by Galectin-3 via TLR4.
Traumatic injury of CNS that encompasses devastation of various neuronal structures, including axonal damage and destruction of myelin, is followed by infiltration of damaged tissue with immune cells and consequent activation of microglia, which precedes regeneration and scarring. In mouse model of cortical contusion, expression of Galectin-3 significantly increased after impact in cortex and hippocampus, followed by increased levels of Galectin-3 in cerebrospinal fluid, and interaction between Galectin-3 and TLR4 [72]. Application of anti-Gal-3 antibody exhibited neuroprotective effect due to decreased trauma-induced synthesis of pro-inflammatory markers, IL-1β, IL-6 and iNOS, and attenuated microglial activation. Conversely, Galectin-3 deletion was unsuccessful in modulation of traumatic brain injury (TBI) induced pro-inflammatory response, suggesting the importance of difference in relation to complete, constitutive absence of Galectin-3 and antagonizing only extracellular Galectin-3. Clinical study on Galectin-3 as a possible prognostic marker in patients with severe TBI showed significant increase of plasma Galectin-3 in TBI patients. The value of Galectin-3 positively correlated with severity noted by Glasgow Coma Scale scores and levels of plasma C-reactive protein [73]. Increase of plasma Galectin-3 was also shown in patients with mild TBI by others [74].
Spinal cord injury (SCI) is also followed by neuroinflammation with significant and prolonged expression of pro-inflammatory proteins, including Galectin-3 [75][76]. Confirming these results, Galectin-3-deficient mice had better functional recovery after SCI due to limitation of lesions without spreading to the surrounding area and maintenance of white matter integrity [77]. With respect to inflammatory response there was a decrease of CD11b-positive cells in Galectin-3-deficient mice, associated with increase of Arginase1-positive cells. Specifically, Arginase1-expressing cells represent anti-inflammatory M2 macrophages characterized by production of anti-inflammatory cytokines [78].
References
- Sciacchitano, S.; Lavra, L.; Morgante, A.; Ulivieri, A.; Magi, F.; De Francesco, G.P.; Bellotti, C.; Salehi, L.B.; Ricci, A. Galectin-3: One Molecule for an Alphabet of Diseases, from A to Z. Int. J. Mol. Sci. 2018, 19, 379.
- Krześlak, A.; Lipińska, A. Galectin-3 as a multifunctional protein. Cell. Mol. Biol. Lett. 2004, 9, 305–328.
- Nio–Kobayashi, J. Tissue– and cell–specific localization of galectins, β–galactose–binding animal lectins, and their potential functions in health and disease. Anat. Sci. Int. 2017, 92, 25–36.
- Dumic, J.; Dabelic, S.; Flögel, M. Galectin-3: An open–ended story. Biochim. Biophys. Acta 2006, 1760, 616–635.
- Birdsall, B.; Feeney, J.; Burdett, I.D.; Bawumia, S.; Barboni, E.A.; Hughes, R.C. NMR solution studies of hamster galectin–3 and electron microscopic visualization of surface–adsorbed complexes: Evidence for interactions between the N– and C–terminal domains. Biochemistry 2001, 40, 4859–4866.
- Mazurek, N.; Conklin, J.; Byrd, J.C.; Raz, A.; Bresalier, R.S. Phosphorylation of the beta–galactoside–binding protein galectin–3 modulates binding to its ligands. J. Biol. Chem. 2000, 275, 36311–36315.
- Huflejt, M.E.; Turck, C.W.; Lindstedt, R.; Barondes, S.H.; Leffler, H. L–29, a soluble lactose–binding lectin, is phosphorylated on serine 6 and serine 12 in vivo and by casein kinase I. J. Biol. Chem. 1993, 268, 26712–26718.
- Flores–Ibarra, A.; Vértesy, S.; Medrano, F.J.; Gabius, H.J.; Romero, A. Crystallization of a human galectin–3 variant with two ordered segments in the shortened N–terminal tail. Sci. Rep. 2018, 8, 1–11.
- Fortuna–Costa, A.; Gomes, A.M.; Kozlowski, E.O.; Stelling, M.P.; Pavão, M.S. Extracellular galectin–3 in tumor progression and metastasis. Front. Oncol. 2014, 4, 138–147.
- Van den Brûle, F.A.; Fernandez, P.L.; Buicu, C.; Liu, F.T.; Jackers, P.; Lambotte, R.; Castronovo, V. Differential expression of galectin–1 and galectin–3 during first trimester human embryogenesis. Dev. Dyn. 1997, 209, 399–405.
- Joo, H.G.; Goedegebuure, P.S.; Sadanaga, N.; Nagoshi, M.; von Bernstorff, W.; Eberlein, T.J. Expression and function of galectin–3, a beta–galactoside–binding protein in activated T lymphocytes. J. Leukoc. Biol. 2001, 69, 555–564.
- Di Lella, S.; Sundblad, V.; Cerliani, J.P.; Guardia, C.M.; Estrin, D.A.; Vasta, G.R.; Rabinovich, G.A. When galectins recognize glycans: From biochemistry to physiology and back again. Biochemistry 2011, 50, 7842–7857.
- Liu, F.T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41.
- Hisrich, B.V.; Young, R.B.; Sansone, A.M.; Bowens, Z.; Green, L.J.; Lessey, B.A.; Blenda, A.V. Role of Human Galectins in Inflammation and Cancers Associated with Endometriosis. Biomolecules 2020, 10, 230.
- Wang, J.L.; Gray, R.M.; Haudek, K.C.; Patterson, R.J. Nucleocytoplasmic lectins. Biochim. Biophys. Acta 2004, 1673, 75–93.
- Hughes, R.C. Secretion of the galectin family of mammalian carbohydrate–binding proteins. Biochim. Biophys. Acta 1999, 1473, 172–185.
- Théry, C.; Boussac, M.; Véron, P.; Ricciardi–Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell–derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318.
- Mehul, B.; Hughes, R.C. Plasma membrane targetting, vesicular budding and release of galectin 3 from the cytoplasm of mammalian cells during secretion. J. Cell. Sci. 1997, 110, 1169–1178.
- Welton, J.L.; Khanna, S.; Giles, P.J.; Brennan, P.; Brewis, I.A.; Staffurth, J.; Mason, M.D.; Clayton, A. Proteomics analysis of bladder cancer exosomes. Mol. Cell. Proteom. 2010, 9, 1324–1338.
- Bänfer, S.; Schneider, D.; Dewes, J.; Strauss, M.T.; Freibert, S.A.; Heimerl, T.; Maier, U.G.; Elsässer, H.P.; Jungmann, R.; Jacob, R. Molecular mechanism to recruit galectin–3 into multivesicular bodies for polarized exosomal secretion. Proc. Natl. Acad. Sci. USA 2018, 115, 4396–4405.
- Arsenijevic, A.; Milovanovic, M.; Milovanovic, J.; Stojanovic, B.; Zdravkovic, N.; Leung, P.S.; Liu, F.T.; Gershwin, M.E.; Lukic, M.L. Deletion of Galectin-3 Enhances Xenobiotic Induced Murine Primary Biliary Cholangitis by Facilitating Apoptosis of BECs and Release of Autoantigens. Sci. Rep. 2016, 6, 1–11.
- Arsenijevic, A.; Milovanovic, J.; Stojanovic, B.; Djordjevic, D.; Stanojevic, I.; Jankovic, N.; Vojvodic, D.; Arsenijevic, N.; Lukic, M.L.; Milovanovic, M. Gal–3 Deficiency Suppresses Novosphyngobium aromaticivorans Inflammasome Activation and IL–17 Driven Autoimmune Cholangitis in Mice. Front. Immunol. 2019, 10, 1–18.
- Kovacevic, M.M.; Pejnovic, N.; Mitrovic, S.; Jovicic, N.; Petrovic, I.; Arsenijevic, N.; Lukic, M.L.; Ljujic, B. Galectin-3 deficiency enhances type 2 immune cell–mediated myocarditis in mice. Immunol. Res. 2018, 66, 491–502.
- Yoo, H.I.; Kim, E.G.; Lee, E.J.; Hong, S.Y.; Yoon, C.S.; Hong, M.J.; Park, S.J.; Woo, R.S.; Baik, T.K.; Song, D.Y. Neuroanatomical distribution of galectin–3 in the adult rat brain. J. Mol. Histol. 2017, 48, 133–146.
- Comte, I.; Kim, Y.; Young, C.C.; van der Harg, J.M.; Hockberger, P.; Bolam, P.J.; Poirier, F.; Szele, F.G. Galectin-3 maintains cell motility from the subventricular zone to the olfactory bulb. J. Cell. Sci. 2011, 124, 2438–2447.
- Partridge, E.A.; Le Roy, C.; Di Guglielmo, G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of cytokine receptors by Golgi N–glycan processing and endocytosis. Science 2004, 306, 120–124.
- Liu, W.; Hsu, D.K.; Chen, H.Y.; Yang, R.Y.; Carraway, K.L., 3rd; Isseroff, R.R.; Liu, F.T. Galectin-3 regulates intracellular trafficking of EGFR through Alix and promotes keratinocyte migration. J. Invest. Dermatol. 2012, 132, 2828–2837.
- Pasquini, L.A.; Millet, V.; Hoyos, H.C.; Giannoni, J.P.; Croci, D.O.; Marder, M.; Liu, F.T.; Rabinovich, G.A.; Pasquini, J.M. Galectin-3 drives oligodendrocyte differentiation to control myelin integrity and function. Cell Death Differ. 2011, 18, 1746–1756.
- Thomas, L.; Pasquini, L.A. Galectin-3–Mediated Glial Crosstalk Drives Oligodendrocyte Differentiation and (Re)myelination. Front. Cell. Neurosci. 2018, 12, 297–313.
- Lee, A.C.; Kozuki, N.; Blencowe, H.; Vos, T.; Bahalim, A.; Darmstadt, G.L.; Niermeyer, S.; Ellis, M.; Robertson, N.J.; Cousens, S.; et al. Intrapartum–related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr. Res. 2013, 74, 50–72.
- Schiering, I.A.; de Haan, T.R.; Niermeijer, J.M.; Koelman, J.H.; Majoie, C.B.; Reneman, L.; Aronica, E. Correlation between clinical and histologic findings in the human neonatal hippocampus after perinatal asphyxia. J. Neuropathol. Exp. Neurol. 2014, 73, 324–334.
- Vlassaks, E.; Brudek, T.; Pakkenberg, B.; Gavilanes, A.W. Cerebellar cytokine expression in a rat model for fetal asphyctic preconditioning and perinatal asphyxia. Cerebellum 2014, 13, 471–478.
- Torres–Cuevas, I.; Corral–Debrinski, M.; Gressens, P. Brain oxidative damage in murine models of neonatal hypoxia/ischemia and reoxygenation. Free Radic. Biol. Med. 2019, 142, 3–15.
- Lalancette–Hébert, M.; Swarup, V.; Beaulieu, J.M.; Bohacek, I.; Abdelhamid, E.; Weng, Y.C.; Sato, S.; Kriz, J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci. 2012, 32, 10383–10395.
- Rahimian, R.; Béland, L.C.; Kriz, J. Galectin-3: Mediator of microglia responses in injured brain. Drug. Discov. Today 2018, 23, 375–381.
- Doverhag, C.; Hedtjärn, M.; Poirier, F.; Mallard, C.; Hagberg, H.; Karlsson, A.; Sävman, K. Galectin-3 contributes to neonatal hypoxic–ischemic brain injury. Neurobiol. Dis. 2010, 38, 36–46.
- Doverhag, C.; Keller, M.; Karlsson, A.; Hedtjarn, M.; Nilsson, U.; Kapeller, E.; Sarkozy, G.; Klimaschewski, L.; Humpel, C.; Hagberg, H.; et al. Pharmacological and genetic inhibition of NADPH oxidase does not reduce brain damage in different models of perinatal brain injury in newborn mice. Neurobiol. Dis. 2008, 31, 133–144.
- Pesheva, P.; Kuklinski, S.; Biersack, H.J.; Probstmeier, R. Nerve growth factor–mediated expression of galectin–3 in mouse dorsal root ganglion neurons. Neurosci. Lett. 2000, 293, 37–40.
- Pesheva, P.; Kuklinski, S.; Schmitz, B.; Probstmeier, R. Galectin-3 promotes neural cell adhesion and neurite growth. J. Neurosci. Res. 1998, 54, 639–654.
- Kuklinski, S.; Vladimirova, V.; Waha, A.; Kamata, H.; Pesheva, P.; Probstmeier, R. Expression of galectin–3 in neuronally differentiating PC12 cells is regulated both via Ras/MAPK–dependent and –independent signalling pathways. J. Neurochem. 2003, 87, 1112–1124.
- Umekawa, T.; Osman, A.M.; Han, W.; Ikeda, T.; Blomgren, K. Resident microglia, rather than blood–derived macrophages, contribute to the earlier and more pronounced inflammatory reaction in the immature compared with the adult hippocampus after hypoxia–ischemia. Glia 2015, 63, 2220–2230.
- Chip, S.; Fernández–López, D.; Li, F.; Faustino, J.; Derugin, N.; Vexler, Z.S. Genetic deletion of galectin–3 enhances neuroinflammation, affects microglial activation and contributes to sub–chronic injury in experimental neonatal focal stroke. Brain. Behav. Immun. 2017, 60, 270–281.
- Al–Dalahmah, O.; Campos Soares, L.; Nicholson, J.; Draijer, S.; Mundim, M.; Lu, V.M.; Sun, B.; Tyler, T.; Adorján, I.; O’Neill, E.; et al. Galectin-3 modulates postnatal subventricular zone gliogenesis. Glia 2020, 68, 435–450.
- Radosavljevic, G.; Volarevic, V.; Jovanovic, I.; Milovanovic, M.; Pejnovic, N.; Arsenijevic, N.; Hsu, D.K.; Lukic, M.L. The roles of Galectin-3 in autoimmunity and tumor progression. Immunol. Res. 2012, 52, 100–110.
- Stajic, D.; Selakovic, D.; Jovicic, N.; Joksimovic, J.; Arsenijevic, N.; Lukic, M.L.; Rosic, G. The role of galectin–3 in modulation of anxiety state level in mice. Brain. Behav. Immun. 2019, 78, 177–187.
- Shin, T. The pleiotropic effects of galectin–3 in neuroinflammation: A review. Acta. Histochem. 2013, 115, 407–411.
- Steinman, L.; Zamvil, S.S. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann. Neurol. 2006, 60, 12–21.
- Jiang, H.R.; Al Rasebi, Z.; Mensah–Brown, E.; Shahin, A.; Xu, D.; Goodyear, C.S.; Fukada, S.Y.; Liu, F.T.; Liew, F.Y.; Lukic, M.L. Galectin-3 deficiency reduces the severity of experimental autoimmune encephalomyelitis. J. Immunol. 2009, 182, 1167–1173.
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643.
- Reichert, F.; Rotshenker, S. Galectin-3/MAC–2 in experimental allergic encephalomyelitis. Exp. Neurol. 1999, 160, 508–514.
- Rotshenker, S.; Reichert, F.; Gitik, M.; Haklai, R.; Elad–Sfadia, G.; Kloog, Y. Galectin-3/MAC–2, Ras and PI3K activate complement receptor–3 and scavenger receptor–AI/II mediated myelin phagocytosis in microglia. Glia 2008, 56, 1607–1613.
- Rotshenker, S. The role of Galectin-3/MAC–2 in the activation of the innate–immune function of phagocytosis in microglia in injury and disease. J. Mol. Neurosci. 2009, 39, 99–103.
- Rotshenker, S. Wallerian degeneration: The innate–immune response to traumatic nerve injury. J. Neuroinflam. 2011, 8, 109.
- Mietto, B.S.; Jurgensen, S.; Alves, L.; Pecli, C.; Narciso, M.S.; Assunção-Miranda, I.; Villa–Verde, D.M.; de Souza Lima, F.R.; de Menezes, J.R.; Benjamim, C.F.; et al. Lack of galectin–3 speeds Wallerian degeneration by altering TLR and pro–inflammatory cytokine expressions in injured sciatic nerve. Eur. J. Neurosci. 2013, 37, 1682–1690.
- Chaudhary, P.; Marracci, G.; Galipeau, D.; Pocius, E.; Morris, B.; Bourdette, D. Lipoic acid reduces inflammation in a mouse focal cortical experimental autoimmune encephalomyelitis model. J. Neuroimmunol. 2015, 289, 68–74.
- Itabashi, T.; Arima, Y.; Kamimura, D.; Higuchi, K.; Bando, Y.; Takahashi–Iwanaga, H.; Murakami, M.; Watanabe, M.; Iwanaga, T.; Nio–Kobayashi, J. Cell– and stage–specific localization of galectin–3, a β–galactoside–binding lectin, in a mouse model of experimental autoimmune encephalomyelitis. Neurochem. Int. 2018, 118, 176–184.
- Mensah–Brown, E.P.; Al Rabesi, Z.; Shahin, A.; Al Shamsi, M.; Arsenijevic, N.; Hsu, D.K.; Liu, F.T.; Lukic, M.L. Targeted disruption of the galectin–3 gene results in decreased susceptibility to multiple low dose streptozotocin–induced diabetes in mice. Clin. Immunol. 2009, 130, 83–88.
- Petrovic, I.; Pejnovic, N.; Ljujic, B.; Pavlovic, S.; Miletic Kovacevic, M.; Jeftic, I.; Djukic, A.; Draginic, N.; Andjic, M.; Arsenijevic, N.; et al. Overexpression of Galectin 3 in Pancreatic β Cells Amplifies β–Cell Apoptosis and Islet Inflammation in Type–2 Diabetes in Mice. Front. Endocrinol. 2020, 11, 1–14.
- Jaquenod De Giusti, C.; Alberdi, L.; Frik, J.; Ferrer, M.F.; Scharrig, E.; Schattner, M.; Gomez, R.M. Galectin-3 is upregulated in activated glia during Junin virus–induced murine encephalitis. Neurosci. Lett. 2011, 501, 163–166.
- James, R.E.; Hillis, J.; Adorján, I.; Gration, B.; Mundim, M.V.; Iqbal, A.J.; Majumdar, M.M.; Yates, R.L.; Richards, M.M.; Goings, G.E.; et al. Loss of galectin–3 decreases the number of immune cells in the subventricular zone and restores proliferation in a viral model of multiple sclerosis. Glia 2016, 64, 105–121.
- Venkatraman, A.; Hardas, S.; Patel, N.; Singh Bajaj, N.; Arora, G.; Arora, P. Galectin-3: An emerging biomarker in stroke and cerebrovascular diseases. Eur. J. Neurol. 2018, 25, 238–246.
- Yan, Y.P.; Lang, B.T.; Vemuganti, R.; Dempsey, R.J. Galectin-3 mediates post–ischemic tissue remodeling. Brain. Res. 2009, 1288, 116–124.
- Rahimian, R.; Lively, S.; Abdelhamid, E.; Lalancette–Hebert, M.; Schlichter, L.; Sato, S.; Kriz, J. Delayed Galectin-3–Mediated Reprogramming of Microglia After Stroke is Protective. Mol. Neurobiol. 2019, 56, 6371–6385.
- Satoh, K.; Niwa, M.; Goda, W.; Binh, N.H.; Nakashima, M.; Takamatsu, M.; Hara, A. Galectin-3 expression in delayed neuronal death of hippocampal CA1 following transient forebrain ischemia, and its inhibition by hypothermia. Brain. Res. 2011, 1382, 266–274.
- Cheng, X.; Boza–Serrano, A.; Turesson, M.F.; Deierborg, T.; Ekblad, E.; Voss, U. Galectin-3 causes enteric neuronal loss in mice after left sided permanent middle cerebral artery occlusion, a model of stroke. Sci. Rep. 2016, 6, 1–8.
- Deierborg, T.; Burguillos, M.A. A new “sweet” ligand for Toll–like receptor 4. Oncotarget 2015, 6, 19928–19929.
- Burguillos, M.A.; Svensson, M.; Schulte, T.; Boza–Serrano, A.; Garcia–Quintanilla, A.; Kavanagh, E.; Santiago, M.; Viceconte, N.; Oliva–Martin, M.J.; Osman, A.M.; et al. Galectin-3 Acts as a Toll–like Receptor 4 Ligand and Contributes to Microglial Activation. Cell. Rep. 2015, 10, 1626–1630.
- Wu, Z.S.; Lo, J.J.; Wu, S.H.; Wang, C.Z.; Chen, R.F.; Lee, S.S.; Chai, C.Y.; Huang, S.H. Early Hyperbaric Oxygen Treatment Attenuates Burn–Induced Neuroinflammation by Inhibiting the Galectin-3–Dependent Toll–Like Receptor–4 Pathway in a Rat Model. Int. J. Mol. Sci. 2018, 19, 2195.
- Stojanovic, B.; Jovanovic, I.; Stojanovic, B.S.; Stojanovic, M.D.; Gajovic, N.; Radosavljevic, G.; Pantic, J.; Arsenijevic, N.; Lukic, M.L. Deletion of Galectin-3 attenuates acute pancreatitis in mice by affecting activation of innate inflammatory cells. Eur. J. Immunol. 2019, 49, 940–946.
- Nomura, K.; Vilalta, A.; Allendorf, D.H.; Hornik, T.C.; Brown, G.C. Activated Microglia Desialylate and Phagocytose Cells via Neuraminidase, Galectin-3, and Mer Tyrosine Kinase. J. Immunol. 2017, 198, 4792–4801.
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.L.; Li, W. Galectin-3 is a new MerTK–specific eat–me signal. J. Cell. Physiol. 2012, 227, 401–407.
- Yip, P.K.; Carrillo–Jimenez, A.; King, P.; Vilalta, A.; Nomura, K.; Chau, C.C.; Egerton, A.M.; Liu, Z.H.; Shetty, A.J.; Tremoleda, J.L.; et al. Galectin-3 released in response to traumatic brain injury acts as an alarmin orchestrating brain immune response and promoting neurodegeneration. Sci. Rep. 2017, 7, 1–13.
- Shen, Y.F.; Yu, W.H.; Dong, X.Q.; Du, Q.; Yang, D.B.; Wu, G.Q.; Zhang, Z.Y.; Wang, H.; Jiang, L. The change of plasma galectin–3 concentrations after traumatic brain injury. Clin. Chim. Acta 2016, 456, 75–80.
- Shan, R.; Szmydynger–Chodobska, J.; Warren, O.U.; Mohammad, F.; Zink, B.J.; Chodobski, A. A New Panel of Blood Biomarkers for the Diagnosis of Mild Traumatic Brain Injury/Concussion in Adults. J. Neurotrauma 2016, 33, 49–57.
- Byrnes, K.R.; Washington, P.M.; Knoblach, S.M.; Hoffman, E.; Faden, A.I. Delayed inflammatory mRNA and protein expression after spinal cord injury. J. Neuroinflam. 2011, 8, 130–141.
- Pajoohesh–Ganji, A.; Knoblach, S.M.; Faden, A.I.; Byrnes, K.R. Characterization of inflammatory gene expression and galectin–3 function after spinal cord injury in mice. Brain. Res. 2012, 1475, 96–105.
- Mostacada, K.; Oliveira, F.L.; Villa–Verde, D.M.; Martinez, A.M. Lack of galectin–3 improves the functional outcome and tissue sparing by modulating inflammatory response after a compressive spinal cord injury. Exp. Neurol. 2015, 271, 390–400.
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399.
More
Information
Subjects:
Neurosciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
866
Revisions:
2 times
(View History)
Update Date:
17 Jun 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No