 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anna Igorevna Sulatskaya | + 1623 word(s) | 1623 | 2021-06-10 08:10:13 | | | |
| 2 | Anna Igorevna Sulatskaya | + 13 word(s) | 1636 | 2021-06-15 06:41:15 | | |
Video Upload Options
Proteolytic enzymes are known to be involved in the formation and degradation of various monomeric proteins, but the effect of proteases on the ordered protein aggregates, amyloid fibrils, which are considered to be extremely stable, remains poorly understood. In this work we study resistance to proteolytic degradation of lysozyme amyloid fibrils with two different types of morphology and beta-2-microglobulun amyloids.
1. Introduction
2. Results and discussion
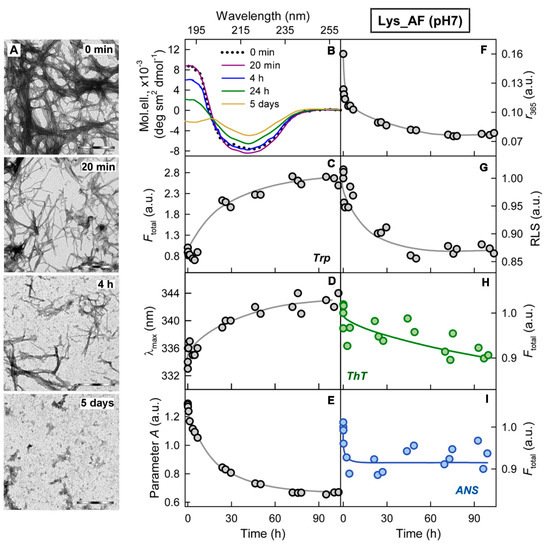
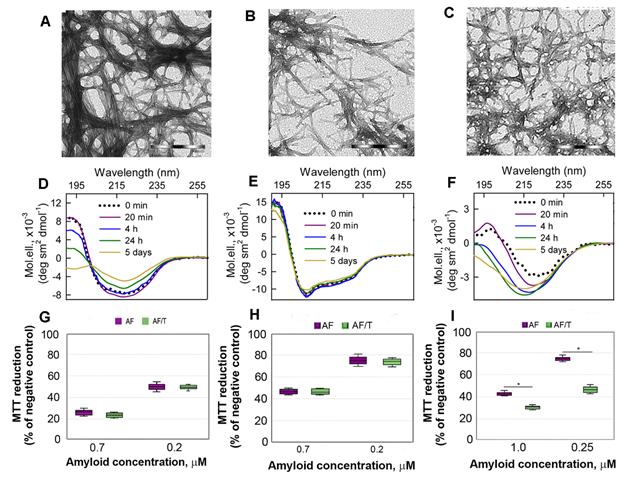
Figure 3. Cytotoxicity of amyloids formed from different proteins. (A-C) The electron micrographs, (D-F) CD spectra and (G-I) dose-dependent inhibition effect on HeLa cells (determined by MTT assay) of amyloid fibrils formed from lysozyme at pH7 (A, D, G) and pH2 (B, E, H) and from beta-2-microglobulin (C, F, I) . Scale bars at the electron micrographs are equal to 1 μm. At the Panels (G-I) data are expressed as medians and interquartile ranges, whiskers denote 1.5 × IQR. ANOVA, Tukey post hoc test, * р < 0.01.
Binding of proteases to amyloids and proteolytic degradation of fibrils, at first glance, seems to be a positive effect in terms of amyloidosis treatment; however, this is actually not entirely true. The interaction of trypsin with abnormal aggregates amyloid fibrils, which was shown in our work, as well as in the work[19], can lead to the inability of the enzyme interaction with other substrates. For example, trypsin is required for Aβ-peptide catabolism, and its interaction with Aβ-peptide amyloids can reduce clearance of soluble Aβ-peptide, and an increased concentration of Aβ-peptide enhances its aggregation. A similar conclusion was reached in the study of the trypsin binding to fibrillar Aβ-peptide[19]. This assumption is also in a good agreement with the results according to which mice deficient of proteolytic enzymes exhibited elevated levels of Aβ while overexpression of neprilysin in APP-transgenic mice could reduce the deposition of fibrillar Aβ[31][32].
In addition, we showed that proteolytic degradation of amyloids did not reduce cytotoxicity of the protein aggregates for Hela cells, as might be expected (at the Figure 3 G-I). We attribute this to the formation of highly toxic non-fibrillar aggregates along with the formation of non-toxic protein monomers and their fragments induced by trypsin. A high cytotoxicity of large disordered aggregates forming after exposure of the lysozyme and beta-2-microglobulin fibrils to the protein with chaperone activity alpha-B-crystallin was already demonstrated by us earlier[33].
In conclusion, we show that the proteolytic enzyme of the pancreas, trypsin, can induce degradation of amyloid fibrils, and the mechanism of this process is qualitatively the same for amyloids with different structure and morphology (Figure 4). At the same time, the efficiency and rate of fibril degradation are dependent on the structure of the amyloid-forming protein as well as on the morphology and clustering of amyloid fibrils, which determines the number, localization, and availability of protease cleavage sites. Proteolytic degradation of amyloids not only does not reduce, as might be expected, but, in some cases, even increases cytotoxicity of the amyloid fibrils, presumably due to the formation of more toxic non-fibrillar aggregates.
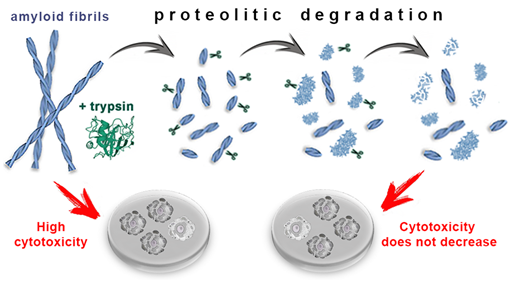
Figure 4. Mechanism of trypsin induced degradation of amyloids formed from different proteins: 1) first, amyloid clots are declustering, and amyloid fibers are fragmented; 2) then, the structure of shortened fibril fragments “decompacts” and becomes less ordered, 3) after which these aggregates degrade to monomers, which, in turn, degrade into short fragments. The cytotoxicity of amyloids treated with trypsin not only does not decrease, but can even increase (this has been demonstrated for beta-2-microglobulin fibrils).
References
- Giampaolo Merlini; Vittorio Bellotti; Molecular Mechanisms of Amyloidosis. New England Journal of Medicine 2003, 349, 583-596, 10.1056/nejmra023144.
- Alessandra Recchia; Patrizia Debetto; Alessandro Negro; Diego Guidolin; Stephen D. Skaper; Pietro Giusti; α‐Synuclein and Parkinson's disease. The FASEB Journal 2004, 18, 617-626, 10.1096/fj.03-0338rev.
- Jun-Xia Lu; Wei Qiang; Wai-Ming Yau; Charles D. Schwieters; Stephen C. Meredith; Robert Tycko; Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 2013, 154, 1257-1268, 10.1016/j.cell.2013.08.035.
- 4.Naiki, H.; Okoshi, T.; Ozawa, D.; Yamaguchi, I.; Hasegawa, K.; Molecular pathogenesis of human amyloidosis: Lessons from beta2 -microglobulin-related amyloidosis. Pathol. Int. 2016, 66, 193, 10.1111/pin.12394.
- Jean D. Sipe; Merrill D. Benson; Joel N. Buxbaum; Shu-Ichi Ikeda; Giampaolo Merlini; Maria J. M. Saraiva; Per Westermark; Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016, 23, 209-213, 10.1080/13506129.2016.1257986.
- G. Esposito; R. Michelutti; G. Verdone; P. Viglino; H. Hern Ández; C. V. Robinson; A. Amoresano; F. Dal Piaz; M. Monti; P. Pucci; et al.P. MangioneM. StoppiniGiampaolo MerliniG. FerriV. Bellotti Removal of the N-terminal hexapeptide from human β2-microglobulin facilitates protein aggregation and fibril formation. Protein Science 2000, 9, 831-845, 10.1110/ps.9.5.831.
- P. T. Sattianayagam; S. D. J. Gibbs; D. Rowczenio; J. H. Pinney; A. D. Wechalekar; J. A. Gilbertson; P. N. Hawkins; H. J. Lachmann; J. D. Gillmore; Hereditary lysozyme amyloidosis - phenotypic heterogeneity and the role of solid organ transplantation. Journal of Internal Medicine 2011, 272, 36-44, 10.1111/j.1365-2796.2011.02470.x.
- Valérie Wilquet; Bart De Strooper; Amyloid-beta precursor protein processing in neurodegeneration. Current Opinion in Neurobiology 2004, 14, 582-588, 10.1016/j.conb.2004.08.001.
- Martijn F. B. G. Gebbink; Emile E. Voest; Arie Reijerkerk; Do antiangiogenic protein fragments have amyloid properties?. Blood 2004, 104, 1601-1605, 10.1182/blood-2004-02-0433.
- Han Zhang; Qilin Ma; Yun-Wu Zhang; Huaxi Xu; Proteolytic processing of Alzheimer’s β-amyloid precursor protein. Journal of Neurochemistry 2011, 120, 9-21, 10.1111/j.1471-4159.2011.07519.x.
- Sebastian Hogl; Peer-Hendrik Kuhn; Alessio Colombo; Stefan F. Lichtenthaler; Determination of the Proteolytic Cleavage Sites of the Amyloid Precursor-Like Protein 2 by the Proteases ADAM10, BACE1 and γ-Secretase. PLOS ONE 2011, 6, e21337, 10.1371/journal.pone.0021337.
- 12. Mirgorodskaia, O.A.; Kazanina, G.A.; Mirgorodskaia, E.P.; Shevchenko, A.A.; Mal'tsev, K.V.; Miroshnikov, A.I.; Roipstorff, P.; [Proteolysis of human proinsulin catalysed by native, modified, and immobilized trypsin]. Bioorganicheskaia Khimiia 1997, 23, 91-97.
- Yu-Zhe Gao; Hong-Hua Xu; Ting-Ting Ju; Xin-Huai Zhao; The effect of limited proteolysis by different proteases on the formation of whey protein fibrils. Journal of Dairy Science 2013, 96, 7383-7392, 10.3168/jds.2013-6843.
- P. Patrizia Mangione; Riccardo Porcari; Julian D. Gillmore; Piero Pucci; Maria Monti; Mattia Porcari; Sofia Giorgetti; Loredana Marchese; Sara Raimondi; Louise C. Serpell; et al.Wenjie ChenAnnalisa ReliniJulien MarcouxInnes R. ClatworthyGraham W. TaylorGlenys A. TennentCarol V. RobinsonPhilip N. HawkinsMonica StoppiniStephen P. WoodMark B. PepysVittorio Bellotti Proteolytic cleavage of Ser52Pro variant transthyretin triggers its amyloid fibrillogenesis. Proceedings of the National Academy of Sciences 2014, 111, 1539-1544, 10.1073/pnas.1317488111.
- Julien Marcoux; P Patrizia Mangione; Riccardo Porcari; Matteo T Degiacomi; Guglielmo Verona; Graham W Taylor; Sofia Giorgetti; Sara Raimondi; Sarah Cianferani; Justin L P Benesch; et al.Ciro CecconiMohsin M NaqviJulian D GillmorePhilip N HawkinsMonica StoppiniCarol V RobinsonMark B PepysVittorio Bellotti A novel mechano‐enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Molecular Medicine 2015, 7, 1337-1349, 10.15252/emmm.201505357.
- Thuzar M Shin; J Mario Isas; Chia-Ling Hsieh; Rakez Kayed; Charles G Glabe; Ralf Langen; Jeannie Chen; Formation of soluble amyloid oligomers and amyloid fibrils by the multifunctional protein vitronectin. Molecular Neurodegeneration 2008, 3, 16-16, 10.1186/1750-1326-3-16.
- Simon Poepsel; Andreas Sprengel; Barbara Sacca; Farnusch Kaschani; Markus Kaiser; Christos Gatsogiannis; Stefan Raunser; Tim Clausen; Michael Ehrmann; Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nature Chemical Biology 2015, 11, 862-869, 10.1038/nchembio.1931.
- Linda Söderberg; Camilla Dahlqvist; Hiroyoshi Kakuyama; Johan Thyberg; Akira Ito; Bengt Winblad; Jan Näslund; Lars O. Tjernberg; Collagenous Alzheimer amyloid plaque component assembles amyloid fibrils into protease resistant aggregates. The FEBS Journal 2005, 272, 2231-2236, 10.1111/j.1742-4658.2005.04647.x.
- Harish Chander; Abha Chauhan; Jerzy Wegiel; Mazhar Malik; Ashfaq Sheikh; Ved Chauhan; Binding of trypsin to fibrillar amyloid beta-protein. Brain Research 2006, 1082, 173-181, 10.1016/j.brainres.2006.01.079.
- Estefania P. C. Azevedo; Anderson B. Guimarães-Costa; Guilherme S. Torezani; Carolina A. Braga; Fernando Palhano; Jeffery W. Kelly; Elvira M. Saraiva; Debora Foguel; Amyloid Fibrils Trigger the Release of Neutrophil Extracellular Traps (NETs), Causing Fibril Fragmentation by NET-associated Elastase*. Journal of Biological Chemistry 2012, 287, 37206-37218, 10.1074/jbc.m112.369942.
- Eugene E. Emeson; Yutaka Kikkawa; Boris Gueft; NEW FEATURES OF AMYLOID FOUND AFTER DIGESTION WITH TRYPSIN. Journal of Cell Biology 1966, 28, 570-577, 10.1083/jcb.28.3.570.
- Ayenachew Bezawork-Geleta; Erica J. Brodie; David A. Dougan; Kaye N. Truscott; LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Scientific Reports 2015, 5, 17397-17397, 10.1038/srep17397.
- Wagner, I.; Arlt, H.; van Dyck, L.; Langer, T.; Neupert, W.; Molecular chaperones cooperate with PIM1 protease in the degradation of misfolded proteins in mitochondria. EMBO J 1994, 13, 5135–5145.
- Satoshi Ninagawa; Kazutoshi Mori; Trypsin Sensitivity Assay to Study the Folding Status of Proteins. BIO-PROTOCOL 2016, 6, e1953, 10.21769/bioprotoc.1953.
- John Kay; A Possible Role for Neutral Proteolysis in the Degradation of Intracellular Proteins. Novartis Foundation Symposia 2008, 75, 219-225, 10.1002/9780470720585.ch14.
- Jose M. Sanchez-Ruiz; Protein kinetic stability. Biophysical Chemistry 2010, 148, 1-15, 10.1016/j.bpc.2010.02.004.
- Jessica Merulla; Elisa Fasana; Tatiana Soldà; Maurizio Molinari; Specificity and Regulation of the Endoplasmic Reticulum-Associated Degradation Machinery. Traffic 2013, 14, 767-777, 10.1111/tra.12068.
- Christopher Pleyer; Jan Flesche; Fahad Saeed; Lysozyme amyloidosis – a case report and review of the literature. Clinical Nephrology. Case Studies 2015, 3, 42-45, 10.5414/cncs108538.
- Roberto Scarpioni; Marco Ricardi; Vittorio Albertazzi; Sara De Amicis; Francesco Rastelli; Lara Zerbini; Dialysis-related amyloidosis: challenges and solutions. International Journal of Nephrology and Renovascular Disease 2016, ume 9, 319-328, 10.2147/ijnrd.s84784.
- Anna I. Sulatskaya; Natalia P. Rodina; Olga I. Povarova; Irina M. Kuznetsova; Konstantin K. Turoverov; Different conditions of fibrillogenesis cause polymorphism of lysozyme amyloid fibrils. Journal of Molecular Structure 2017, 1140, 52-58, 10.1016/j.molstruc.2016.10.037.
- Malcolm A Leissring; Wesley Farris; Alice Y Chang; Dominic M Walsh; Xining Wu; Xiaoyan Sun; Matthew P Frosch; Dennis J Selkoe; Enhanced Proteolysis of β-Amyloid in APP Transgenic Mice Prevents Plaque Formation, Secondary Pathology, and Premature Death. Neuron 2003, 40, 1087-1093, 10.1016/s0896-6273(03)00787-6.
- Robert A. Marr; Edward Rockenstein; Atish Mukherjee; Mark S. Kindy; Louis B. Hersh; Fred H. Gage; Inder M. Verma; Eliezer Masliah; Neprilysin Gene Transfer Reduces Human Amyloid Pathology in Transgenic Mice. The Journal of Neuroscience 2003, 23, 1992-1996, 10.1523/jneurosci.23-06-01992.2003.
- Olga V. Stepanenko; M. I. Sulatsky; E. V. Mikhailova; Olesya V. Stepanenko; O. I. Povarova; I. M. Kuznetsova; K. K. Turoverov; A. I. Sulatskaya; Alpha-B-Crystallin Effect on Mature Amyloid Fibrils: Different Degradation Mechanisms and Changes in Cytotoxicity. International Journal of Molecular Sciences 2020, 21, 7659, 10.3390/ijms21207659.
- Olga V. Stepanenko; M. I. Sulatsky; E. V. Mikhailova; Olesya V. Stepanenko; O. I. Povarova; I. M. Kuznetsova; K. K. Turoverov; A. I. Sulatskaya; Alpha-B-Crystallin Effect on Mature Amyloid Fibrils: Different Degradation Mechanisms and Changes in Cytotoxicity. International Journal of Molecular Sciences 2020, 21, 7659, 10.3390/ijms21207659.

